
Abstract
Poly(ether ketone ketone) (PEKK) is a thermoplastic with excellent performance. To date, most PEKK copolymers are synthesized by a two-step method. However, in this work, a series of PEKK copolymers with diverse T/I (terephthaloyl/isophthaloyl) ratios were firstly synthesized by a facial one-step method. Then, the structure of PEKK copolymers was characterized and the crystallization and melting behavior were studied in detail. With the increase in meta isomers in the structure, the crystallization ability and melting point of copolymers reduced, as confirmed by non-isothermal and isothermal crystallization kinetics analysis. Furthermore, the double melting behavior of PEKK copolymers after isothermal annealing was researched employing differential scanning calorimetry (DSC) and temperature-modulated DSC (TMDSC). Unlike many other outcomes, our study demonstrate that the double melting behavior is mainly related to the superimposition of rigid amorphous devitrification and enthalpy relaxation. Finally, through the self-nucleation experiment of PEKK70 and PEKK80, the ideal self-nucleation temperatures (Ts,ideal) corresponding to the copolymers were determined, then the successive self-nucleation and annealing (SSA) technology was employed to analyze PEKK copolymers for the first time, and thermal classification was successfully achieved.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Poly(aryl ether ketones) (PAEKs) are widely used in the aerospace industry because of the superior high-temperature performance, mechanical properties and corrosion resistance [1,2,3,4,5]. Poly(ether ether ketone) (PEEK) and poly(ether ketone ketone) (PEKK) are both well-known instances in the PAEKs family. Nevertheless, PEKK copolymers are preferred in industrial production and application because of the better economic performance and thermal properties. On one hand, the preparation cost of PEKK copolymers is lower [6, 7], and on the other hand, PEKK copolymers have adjustable crystallization rates and show wider processing windows, making them ideal candidates for composite materials [8].
PEKK was first prepared by Bonner [9] through Friedel–Crafts acylation reaction, unfortunately, the molecular weight of the product was too low to be suitable for practical applications. Subsequently, several endeavors had been made to synthesize high molecular weight PEKK by choosing different solvents or catalysts [10,11,12,13]. In 1989, Gay et al. [14] discovered a two-step route that successfully synthesized PEKK with high molecular weight, thereafter, PEKK has been synthesized mainly by the two-step method in the industry [15].
The crystallization morphology and melting behavior of PEKK copolymers with various T/I ratios were comprehensively studied by DuPont research groups in the early 1990s [16, 17]. The addition of isophthaloyl unit decreased the equilibrium melting point and crystallization rate. When the crystallization conditions or T/I ratio are different, the PEKK crystalline structure may exhibits two different polymorphs (“Form I” and “Form II”). Gardner et al. [18] conducted an in-depth study on the polymorphism of PAEKs, they deemed that the polymorphism mainly depends on two factors: the stiffness of the polymer chain and molecular mobility during the crystallization process.
Multiple melting behavior has been discovered in many semicrystalline polymers, for instance, poly(lactic acid) (PLA) [19,20,21,22,23], poly(ethylene terephthalate) (PET) [24, 25]. In the differential scanning calorimetry (DSC) heating curves after isothermal crystallization, PEKK with T/I ratios of 90/10, 80/20 and 70/30 exhibited apparent double melting behavior. In contrast, PEKK with T/I ratios of 60/40 and 50/50 showed more complex melting behavior [16]. Double melting behavior has also been found in PEEK and has been studied by many researchers [26,27,28,29,30,31,32]. So far, there are two major explanations for the multiple melting behavior of PAEKs. The first is the mechanism of melt-recrystallization, namely, the low-temperature melting peak is caused by the melting of the original crystal lamellae, followed by recrystallization to produce crystals with a higher melting point. The other interpretation of the multiple melting behavior is the presence of various populations of crystals with several melting temperatures. In a word, the controversy still remains in the cause of PAEKs multiple melting behavior.
In recent years, with the commercialization of PEKK and its unique advantages in thermoplastic composites, the research on PEKK copolymers has increased gradually. The corresponding articles mainly focus on crystallization kinetic models [33,34,35], the thermal aging cross-linking degradation [36, 37] and the dynamical mechanical properties [38, 39]. As comprehended from relevant literatures, most PEKK copolymers are derived from the two-step synthesis route. There are few reports on the crystallization and melting behavior of PEKK synthesized by the facile one-step method. Furthermore, successive self-nucleation and annealing (SSA) is currently recognized as a thermal fractionation method and has been adopted by many researchers [40,41,42,43,44,45,46,47,48]. Nevertheless, it is still lacking the exploration of application of SSA technology on PEKK copolymers.
Therefore, in this paper, to determine the relationship between T/I ratio and crystallization melting behavior, we investigated the non-isothermal, isothermal crystallization behavior, spherulite morphology and polymorphism of PEKK copolymers synthesized by a facile one-step method. Regarding the multiple melting behavior of PEKK copolymers after isothermal annealing, the mechanism of multiple melting peaks was discussed from the perspectives of isothermal annealing temperature, isothermal annealing time and temperature-modulated DSC (TMDSC). In addition, the self-nucleation experiments were carried out on PEKK70 and PEKK80 samples, and further SSA experiments were conducted to confirm the feasibility of the SSA method in PEKK copolymers.
Experimental
Materials
The PEKK copolymers are prepared by a facile one-step process employing diphenyl ether (DPE), terephthaloyl chloride (TPC) and isophthaloyl chloride (IPC) as monomers and aluminum chloride (AlCl3) as catalyst. The PEKK copolymers used in this study are provided by Shandong Kaisheng New Materials Co., Ltd. The monomer feed ratios of terephthaloyl chloride with para phenyl links to isophthaloyl chloride with meta phenyl links are 50/50, 60/40, 70/30 and 80/20 for PEKK50, PEKK60, PEKK70 and PEKK80. The intrinsic viscosity of the samples is measured by the Ubbelohde viscometer in 95% sulfuric acid at 30℃. The facile one-step method can achieve intrinsic viscosity values similar to the traditional two-step method [15]. Samples with the intrinsic viscosity around 0.85 dL g−1 (as shown in Table S1) are selected as the reference in order to compare the crystallization properties of PEKK copolymers with different T/I ratios.
Characterizations
Nuclear magnetic resonance (NMR)
The 1H and 13C NMR spectra are measured on a Bruker Avance II 400 M NMR spectrometer and Bruker Avance III 500 M NMR spectrometer, respectively. The samples are dissolved in deuterated trifluoroacetic acid (CF3COOD) solvent and measured at 25 °C.
Differential scanning calorimetry (DSC)
DSC measurements are achieved by employing TA Instruments Q25 in aluminum pans under nitrogen atmosphere.
To investigate the non-isothermal crystallization and consequent melting behavior of PEKK copolymers with various T/I ratios, initially, the samples (about 5 mg) are heated to 380 °C (390 °C for PEKK80) to erase the thermal history. Then, the samples are cooled to 50 °C at 10 °C min−1 and reheated to 380 °C (390 °C for PEKK80) at the same rate. Before and after cooling, the samples are kept in isothermal condition for 3 min.
For the research of isothermal crystallization, the PEKK50, PEKK60 and PEKK70 samples are firstly heated to 380 °C and kept for 3 min, then speedily cooled to the presupposed crystallization temperature (Tc). The isothermal time should be chosen long enough to guarantee complete crystallization of the PEKK copolymers. Finally, the samples are heated immediately from the desired isothermal temperatures at 10 °C min−1.
To study the effect of isothermal time on the appearance of the multiple melting peaks, the PEKK copolymers are processed under the similar molten state, except for the difference of crystallization time. Then the samples are heated instantly from the required isothermal temperatures up to 380 °C at 20 °C min−1.
For the TMDSC experiments, TA Instruments Q25 (with the modulation function) is employed to research the multiple melting behavior of PEKK copolymers. The heating rate is set as 3 °C min−1 with a period of 60 s and a modulation amplitude of 0.5 °C.
Polarized optical microscopy (POM)
The spherulite morphologies of PEKK copolymers are watched by a Leica DM4500P polarizing microscope outfitted with a digital camera. The PEKK copolymers are heated to 380 °C (390 °C for PEKK80) and held for 3 min using Linkam THMS600 hot stage, and then cooled rapidly to the presupposed temperatures. After reaching the temperatures, the spherulite morphologies are recorded at intervals.
Wide-angle X-ray diffraction (WAXD)
WAXD experiment is accomplished on a Dmax-Ultimat X-ray diffractometer (Rigaku, Japan) with Ni-filtered Cu/Kα radiation (λ = 0.15418 nm, 240 kV, 50 mA). Scans are made from 5° to 40° at a rate of 10° min−1. The powder samples are tested after grinding, and the molten crystallized samples are heated to 380 °C (390 °C for PEKK80) to eliminate the thermal history and then crystallized isothermally at 230 °C.
Thermal fractionation by SSA
SSA shall be designed using the ideal self-nucleation temperature (Ts,ideal) as the first Ts,1. Prior to the SSA process, self-nucleation (SN) is carried out to search for Ts,ideal, and the detailed procedure is depicted in Fig. S1. Approximately 3 mg of PEKK70 and PEKK80 samples is employed to complete the SSA tests. The process to employ SSA is presented schematically in Fig. 1, and the detailed steps are described as follows:
-
(a)
The samples are raised to 380 °C for PEKK70 and 390 °C for PEKK80 to erase the thermal history.
-
(b)
Then, the samples are cooled down to 50 °C at 20 °C min−1, and they are held for 3 min in the isothermal state.
-
(c)
Next, the samples are heated at 20 °C min−1 up to the first Ts temperature (Ts,ideal), and they are kept for 5 min in an isothermal state.
-
(d)
Then, the samples are again cooled at 20 °C min−1 down to 50 °C so that the fraction of the samples crystallized during cooling.
-
(e)
Repeated the (c) and (d) steps until all set Ts temperatures are completed (the fractionation window of 5 °C is selected).
-
(f)
Finally, the samples are heated at 20 °C min−1 up to 380 °C for PEKK70 and 390 °C for PEKK80. The final heating scans are performed to melt the previously produced thermal fractions.

Schematic diagram of thermal classification program
Results and discussion
Structure characterization of PEKK copolymers
The structure and composition of PEKK copolymers were determined by 1H NMR spectroscopy as shown in Fig. 2. The positions 1, 2 and 3 show the peaks associated with the IPC links, while the peak corresponding to TPC unit is located at position 4. The chemical environment of H6 and H7 is similar, and the peaks at position 6 and position 7 overlap (the same applies to H4 and H5). The T/I ratio in the structure of PEKK copolymers can be determined by comparing the proportional relationship between the integration of the peak connected with the H1 or H2 and the H6,7 [49].

1H NMR spectrum of PEKK copolymers
Let \(x\) be the fraction of “T” unit, \(y\) be the fraction of “I” unit. When setting the integration value of H6,7 equal to 4, then:
where \(\int {H}_{1}\) and \(\int {H}_{2}\) are the integration of the H1 and H2 resonance peaks. The actual feeding ratio of TPC and IPC and the T/I values calculated by 1H NMR spectrum are listed in Table 1.
To further verify the structure of PEKK copolymers, 13C NMR was also employed. As shown in Fig. 3, the signals from the carbonyl group, at 200.0–200.5 ppm, matching to the T and I units. It can be found that the intensity of the peaks associated with the I units increases with the rise of the amount of IPC in the monomer feed. The ratio of T/I calculated by integrating the corresponding position peak of a and b, the actual feeding ratio of TPC and IPC and the T/I values calculated by 13C NMR spectrum are also recorded in Table 1. It is noteworthy that the 1H and 13C NMR spectra of PEKK copolymers obtained by the one-step method are actually the same as those obtained by the two-step method [15, 49]. Therefore, it is hard to ascertain the sequence structure of different routes through NMR spectrum.

a Chemical structure and b 13C NMR spectra of PEKK copolymers. c Enlarged region of 105.0–120.0 ppm, d enlarged region of 125.0–145.0 ppm, e enlarged region of 160.0–163.0 ppm, f enlarged region of 199.5–201.0 ppm
Non-isothermal crystallization and melting behavior
The thermal properties are closely linked to the actual use of polymers. Therefore, DSC was employed to research the non-isothermal crystallization and subsequent melting behavior of PEKK copolymers with different T/I ratios. The DSC curves of PEKK copolymers are shown in Fig. 4. In detail, the parameters of interest are listed in Table 2. Among them, the values of crystallinity (Xc) are calculated from Eq. 3, where ΔH100% = 130 J g−1 is the melting enthalpy of 100% crystalline PEKK, the same as that of PEEK [50].

a Cooling DSC curves after eliminating the thermal history. b Subsequent heating DSC curves of PEKK copolymers. c Changes in Tg and Tm as a function of the T unit molar fraction and comparison with the two-step method
It is clearly noticed that PEKK50 presents extremely weak crystallization ability. No crystallization peak can be observed in the cooling curve, and a cold crystallization peak appears in the following heating scan. With the increasing of T content, both the glass transition temperature (Tg) and the melting point (Tm) of PEKK tended to increase, but the Tg increased relatively slowly (the Tg of PEKK60-80 are maintained at about 166 °C). The variation of Tg and Tm in the PEKK copolymers with the molar fraction of the T unit and comparison with the two-step method is shown in Fig. 4c. These data suggested that the crystallinity and crystallization ability are enhanced with the increase in T content (the crystallinity raised from 1.8% of PEKK50 to 28.8% of PEKK80). The reason for the above phenomenon is that the addition of meta isomers introduces defects, which reduces the melting temperature and the crystallization rate; meanwhile, the effect is more significant with the increase in the meta isomers content. It can be concluded that PEKK copolymers have tunable crystallization rates and a wider processing window can be obtained by adjusting the T/I ratio. It is worth mentioning that the PEKK50/50 copolymers, synthesized by the two-step method, have two melting peaks during the heating process, located at 305 °C and 331 °C, respectively [16]. In addition, the Tg and Tm of the one-step synthesized PEKK copolymers are higher than those of the two-step synthesized PEKK copolymers, and as the increase in the content of terephthaloyl, the Tg and Tm of the PEKK copolymers synthesized by the two synthesis methods become closer.
Isothermal crystallization
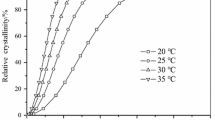
It is crucial to achieve isothermal crystallization in preheating molds for PEKK products with high heat resistance requirements. Thus, the isothermal crystallization behavior of PEKK copolymers with a series of T/I ratios was explored. The DSC exotherms of isothermal crystallization for PEKK50 in the temperature range of 210–250 °C are presented in Fig. 5a. According to the formula of relative crystallinity (Eq. 4), it can be obtained that conversion curves of relative crystallinity (Xt) which are shown in Fig. 5b. In the curves of time and relative crystallinity, as isothermal crystallization temperature (Tc) from 210 °C to 250 °C, the time required to reach the same crystallinity first decreased and then increased.

a Heat flow curves of isothermal crystallization. b Relative crystallinity versus time curves. c The Avrami plots for PEKK50 copolymer crystallized isothermally at 210–250 °C
The reason for this phenomenon is that the diffusion ability of the molecular chain is enhanced as the Tc rises in the low-temperature range, so the crystallization rate increases gradually. However, with a further increase in crystallization temperature, the movement of the molecular chain becomes extremely active and it becomes difficult to form stable crystal nuclei, which causes the crystallization rate to increase first and then decrease with the rise of crystallization temperature.
The Avrami Eq. (5) was used to investigate the isothermal crystallization behavior of PEKK copolymers, and its logarithmic form can be expressed as (Eq. 6). The plots (by plotting \(\text{ln}\left[-\text{ln}\left(1-{X}_{t}\right)\right]\) against lnt) for the PEKK50 copolymer isothermally crystallized in the temperature range of 210–250 °C are shown in Fig. 5c. In order to minimize the influence of secondary crystallization on the isothermal crystallization kinetic parameters [33,34,35], data with relative crystallinity of 30%-70% were selected for fitting. Similarly, the corresponding curves of PEKK60 are shown in Fig. S2. For PEKK70 copolymer, due to its high crystallization ability, only its isothermal crystallization in the high-temperature region has been studied, and the results are shown in Fig. S3. The parameters related to isothermal crystallization include n, k and half-time (t1/2) (the time required to reach half of the total crystallinity) are recorded in Table 3.
For the PEKK50 copolymer, the Avrami exponent (n) has been calculated at 2.38–2.68, revealing a crystal growth of three-dimensional spherulite morphology with heterogeneous nucleation. However, the n values of PEKK60 and PEKK70 are closer to 2, reflecting a two-dimensional crystallization growth with instantaneous nucleation, which is not consistent with the spherulite structure of PEKK observed by polarized optical microscopy. The reason is that, it is difficult to record the complete crystallization peak employing DSC at the onset of isothermal crystallization due to the thermal inertia effects (the thermal inertia of the instrument suddenly changes from the cooling to isothermal step) [51]. The higher the crystallization rate, the more obvious the gap between the crystallization peak and the baseline (as shown in Fig. S4). For incomplete crystallization peak, the starting point of isothermal crystallization is determined by the intersection of linear extrapolation and the base line. According to the research of Jonas et al. [51], the selection of the crystallization origin point influenced the Avrami value by more than 10%.
The peak-time method (the time when the crystallization exothermic peak reaches its maximum) is another way to study isothermal crystallization, which is less affected by the gap at the onset of isothermal crystallization. The isothermal crystallization process of PEKK copolymers synthesized by one-step method was characterized by the peak-time method and compared with the PEKK copolymers synthesized by the two-step method reported in the literature [16], and the results are presented in Fig. 6.

Comparison of peak time for PEKK copolymers synthesized by one-step method with PEKK copolymers synthesized by two-step method reported in the literature [16]
The larger the peak time during isothermal crystallization, the slower the crystallization rate. It is clearly detected that the crystallization rate of PEKK copolymer synthesized by one-step method increased with the decrease in isophthalate content. This is consistent with the results above. Nevertheless, when the PEKK samples were synthesized by the two-step method, samples with a ratio of 50/50 had a higher crystallization rate than those with a ratio of 60/40. It is different from the above experimental results that the samples were synthesized by a facile one-step method. This is due to the fact that the PEKK50/50 sample, employing the two-step synthesis method, is equivalent to the homopolymer with TI diads. Although it has a high content of isophthalate, it still exhibits a higher crystallization rate. However, the PEKK60/40 copolymer also has a high content of isophthalate, and its molecular structure is more random, so it exhibits a slower crystallization rate. Another difference between the one-step mothed and the two-step method is that the copolymers of PEKK50 and PEKK60 have the highest crystallization rate at 230 °C. However, the copolymers of PEKK50/50 and PEKK60/40 have the highest crystallization rate at 250 °C. From these results, it can be noticed that samples synthesized by one-step method have more advantages in crystallization at low temperatures.
The Hoffman-Weeks method was widely employed to evaluate the equilibrium melting point (\({T}_{\rm m}^{0}\)) [52] due to its simplicity. The connection between melting and isothermal crystallization temperature is shown in Fig. 7, the \({T}_{\rm m}^{0}\) reached 367, 348, 324 and 315 °C for the PEKK80, PEKK70, PEKK60 and PEKK50, respectively. This means that melt processing may require higher processing temperatures with increasing T content.

Equilibrium melting obtained by the Hoffman-Weeks method
Spherulite morphology
The polarized optical microscopy (POM) enabled intuitive observation of the growth and morphology of crystalline polymer crystals. POM was applied to observe the morphology of PEKK copolymers generated during isothermal crystallization under the same undercooling temperature. The POM photographs of PEKK copolymers are presented in Fig. 8, at the same time, the spherulite growth process of PEKK50 and PEKK60 are shown in Figs. S5 and S6, respectively.

POM photographs for the samples isothermally crystallized under the same undercooling temperature a PEKK50 at 271 °C, b PEKK60 at 290 °C, c PEKK70 at 314 °C, d PEKK80 at 333 °C
The PEKK50 copolymer has a slower crystallization rate and a smaller number of nuclei are observed under the polarized optical microscopy, with the extension of isothermal time, the volume of spherulites increases gradually and larger spherulites can be formed when they collide with each other. With the rise of T content, the number of nuclei increases; however, the diameters of spherulites decrease when they collide with each other. As for PEKK70, the size of the spherulites decreases sharply, and densely packed crystals with blurred boundaries are observed. The crystal nucleus density changes drastically upon further increase in the T content (PEKK80); meanwhile, finer crystals are observed. All these phenomena show that the crystallization rate and crystallinity gradually enhance with the rise of T unit, which is consistent with the DSC data above. In addition, the spherulite morphology observed is not consistent with the Avrami exponent, and the possible reasons have been discussed in the isothermal crystallization part.
Polymorphism
The crystalline form of PEKK copolymers of unmelt-treated powder and melt-crystallized samples was investigated by WAXD. The diffraction pattern of PEKK after melting and isothermal crystallization at 230℃ is shown in Fig. 9a. There are three diffraction peaks at 2θ = 18.7°, 2θ = 20.6° and 2θ = 22.7° corresponding to the (110), (111) and (200) crystal planes of PEKK, respectively [7, 16, 53]. These diffraction peaks are typical of the “Form I” structure which is consistent with the diffraction patterns of PEEK. The diffraction curves of as-synthesized PEKK powder without melting treatment are presented in Fig. 9b. A different diffraction pattern was observed in the unmelt-treated powder samples, which differed from the “Form I” diffraction peak by a new peak at 2θ = 16° that was related to the “Form II” structure, and this peak corresponds to the (010) reflection of the “Form II” structure [18].

WAXD patterns of a PEKK copolymers after melting and isothermal crystallization at 230℃, b as-synthesized PEKK copolymers powder without melting treatment
It will not affect the crystalline form that the PEKK copolymers synthesized by the one-step or two-step method. Interestingly, the melt-crystallized and as-synthesized samples exhibited different crystal forms. The reason is that the polymorphism of poly aryl ether ketones (PAEKs) is related to the molecular mobility during the crystallization process. Under the condition of melt crystallization, the molecular chain has stronger movement ability, at the moment, diffraction curves are characteristic of the “Form I” structure. Furthermore, for the powder samples that have not been fused, their synthesis and post-processing are conducted at lower temperatures (well below the melting point). The molecular chain activity is limited, which results in a “Form II” structure of the diffraction curves, which were consistent with the results from previous studies [18].
Multiple melting behavior
Although PEKK has two crystal modifications, the WAXD pattern indicated only “Form I” modification under melt crystallization. Furthermore, it has been shown in the literature that the crystal “Form I” is more stable than the crystal “Form II” [18], consequently, under our experimental conditions, there will be no change in crystal form, which can rule out the possibility that multiple melting peaks arise from different crystal modifications.
The crystallization temperature is an essential factor affecting the multiple melting behavior. The DSC heating scan curves of PEKK copolymers after isothermal crystallization at different temperatures (Tc) are displayed in Fig. 10. The melting peak temperature values are listed in Table S2. It is obvious that the temperatures of low-temperature melting peaks increase with the rise of the Tc, and the low-temperature melting peaks occur 10–20℃ above the Tc, such behavior also discovered in PEEK. In addition, the high-temperature melting peaks increase slightly with the increase in the Tc, which is related to the lamella thickening. It is worth noting that the fitted curve of the temperatures of low-temperature melting peaks versus Tc is almost parallel to the line of Tm = Tc, which is a typical characteristic of the annealing peak (as shown in Fig. S7).

DSC heating scans for a PEKK50, b PEKK60, c PEKK70, d PEKK80 after isothermally melt-crystallized at indicated temperatures
Further research was conducted on the effect of crystallization time on the multiple melting behavior of PEKK copolymer. The DSC heating curves of PEKK60 samples after isothermal crystallization for different times are shown in Fig. 11. It is discovered that PEKK60 copolymer could not complete crystallization within 2 min at 230 °C, which was confirmed by a cold crystallization peak that appeared during the subsequent heating. With the extension of isothermal time, the crystallinity increases in the isothermal state, therefore the cold crystallization peak gradually weakens. The same phenomenon also occurs when the Tc at 250 °C. When the isothermal crystallization time exceeds the half-time (t1/2) (the corresponding values are shown in Table 3), the low-temperature melting peaks are gradually obvious. The factors affecting the low-temperature melting peaks may mainly occur after the isothermal crystallization time exceeds half crystallization. This is also reflected in the PEKK50 and PEKK70 samples. The DSC heating scans of the PEKK50 and PEKK70 samples crystallized at different isothermal times are shown in Figs. S8 and S9.

The subsequent DSC heating scan curves of PEKK60 at a 230 °C and b 250 °C isothermal at different times
Temperature-modulated DSC (TMDSC) superimposes a sinusoidally modulated temperature on the conventional linear heating rate, thereby separating the reversing and non-reversing heat flow signals in the total heat flow. TMDSC can provide richer and more accurate information for the complex melting behavior. The multiple melting behaviors of PEKK copolymers were investigated using TMDSC. The TMDSC curve of PEKK70 after isothermal crystallization at 250 °C and 290 °C for 40 min is presented in Fig. 12. It shows typical double melting peaks in the total heat flow curves; meanwhile, in the reversible heat flow curves, a process similar to glass transition appears (marked in yellow in Fig. 12) at the position of the low-temperature melting peak, which is actually the process of rigid amorphous fraction transformed into a mobile amorphous fraction. The rigid amorphous phases have been found and studied in other polymers [54,55,56,57,58,59,60]. The reason why the rigid amorphous phase shows higher transition temperature than the amorphous phase is that the movement of its molecular chains is restrained by the adjacent lamella, resulting in poor chain mobility. In the non-reversible heat flow curve, an endothermic peak also appeared at the position of the low-temperature melting peak, which corresponds to the enthalpy relaxation of the rigid amorphous phase [55, 59]. The same phenomenon is also observed in PEKK50, PEKK60 and PEKK80, and the corresponding TMDSC curves are shown in Figs. S10-S12, respectively. These results denote that the low-temperature melting peaks are the annealing peaks. The double melting behavior is mainly related to the superposition of devitrification and enthalpy relaxation of the rigid amorphous fractions, rather than the melting and recrystallization of the original crystal lamellae as described by the melt-recrystallization mechanism.

TMDSC curve of PEKK70 after isothermal crystallization a at 250℃ and b 290℃ for 40 min
Successive self-nucleation and annealing (SSA)
The SSA technology utilizes the heterogeneous nature of polymer chains to achieve thermal fractionation of polymers through continuous self-nucleation and annealing, which is a means to rapidly achieve the physical fractionation of polymers. It is necessary to perform SN to determine the ideal self-nucleation temperature (Ts,ideal) before the SSA procedure. The self-nucleation technique was applied to the PEKK70 and PEKK80. The DSC scans from each indicated Ts are shown in Fig. 13. From the cooling curves, it can be observed that the position and size of the crystallization peak remain unchanged in the subsequent cooling process at Ts ≥ 368 °C for PEKK70 or Ts ≥ 381 °C for PEKK80. Thus, it is deemed that the sample is in the complete melting domain (Domain I). At 359 °C ≤ Ts ≤ 367 °C for PEKK70 or 375 °C ≤ Ts ≤ 380 °C for PEKK80, the exothermic crystallization peak temperature rises and the peak area decreases compared with the complete melting domain, suggesting the crystallization capacity of the polymer is enhanced during cooling from the melt; meanwhile, no annealing peaks were found in the consequent heating curves. This state represents the self-nucleation domain (Domain II). As Ts continually decreased to 358 °C for PEKK70 or 374 °C for PEKK80, at this time, the temperature was not enough to melt the polymer mostly. Some unmelted crystals annealed within 5 min of isothermal temperature, and additional high-temperature annealing peak appeared in the subsequent heating curves, as discovered in Fig. 13b and d (signaled with arrows). During this process, both the self-nucleation and annealing processes occurred concurrently (Domain III). Therefore, the ideal self-nucleation temperature (Ts,ideal) for PEKK70 and PEKK80 was determined to be 359 °C and 375 °C, respectively, through the above studies. Furthermore, it was found that the self-nucleation temperature ranges of PEKK70 and PEKK80 are 359–367℃ and 375–380℃, respectively.

a DSC cooling curves of PEKK70 after isothermal at Ts and b subsequent heating curves. c DSC cooling curves of PEKK80 after isothermal at Ts and d subsequent heating curves
We investigated an SSA protocol appropriate for fractionating PEKK, and thermal fractionation was applied to PEKK70 and PEKK80 samples. As indicated in the SN results, the first self-nucleation temperature of PEKK70 and PEKK80 samples was chosen to be 359 °C and 375 °C, which represent the Ts,ideal of the PEKK70 and PEKK80 samples, respectively. The final heating curves after thermal fractionation are displayed in Fig. 14, where the thermal fractionation temperature used by the procedure is marked on the graph and divided by a vertical line. The seven fractions obtained for the PEKK70 sample after an eight-step SSA protocol are presented in Fig. 14a, where the Ts,ideal only occurs self-nucleation and no annealing peak appears in the final heating curve. As described in the SN, Ts,2 is in domain III; therefore, the annealing took place within 5 min of isothermal temperature Ts,2, and the polymer self-nucleated in the subsequent cooling. The first thermal fraction is produced in the sample. The same is true at subsequent thermal fraction temperatures. Thus, seven fractions were obtained from PEKK70 samples after the eight-step SSA protocol. Similarly, the seven fractions of the PEKK80 sample after the eight-step SSA protocol are shown in Fig. 14b. The SSA fractionation profiles showed a succession of melting peaks for PEEK70 and PEKK80 associated with the melting of crystals with various average lamella thicknesses generated at severally thermal fractionation temperature Ts (except Ts,ideal). The successful implementation of SSA on PEKK copolymers laid the foundation for subsequent research on the difference of chemical compositions and sequences synthesized by various synthesis methods.

DSC heating curves for a PEKK70 and b PEKK80, before and after SSA fractionation
Conclusions
The crystallization and melting behavior of PEKK copolymers synthesized by a facial one-step method were studied in detail. With the rise of isophthalate unit content, the melting temperature reduces gradually, while the glass transition temperature changes slightly. The isothermal crystallization behavior of PEKK copolymers was investigated by Avrami equation, which has limitations for PEKK copolymers with fast crystallization rate, thereby the isothermal process was analyzed by the peak-time method. The crystallization rate increases with decreasing isophthalate content, which is confirmed by the isothermal crystallization results. It is worth noting that PEKK copolymers synthesized by different methods have distinct crystallization and melting properties. The result is instructive for the performance control of high-performance PEKK materials.
The as-synthesized powdered and the melt-crystallized PEKK copolymers have different crystal forms. This is associated with the chain stiffness and molecular mobility during the crystallization process. Regarding the multiple melting peaks, the fitted curve of the low-temperature melting peaks to the Tc are almost paralleled to the curve of Tm = Tc with the characteristics of annealing peak. Moreover, the low-temperature melting peak occurred when the annealing time exceeded the half-time (t1/2). For the TMDSC results, the endothermic peak in the non-reversible heat flow curve and the glass transition in the reversible heat flow curve confirmed that the low-temperature melting peak originated from the enthalpy relaxation and the transformation of the rigid amorphous phase to the mobile amorphous phase. Finally, the technique of SSA was applied to PEKKs for the first time. The PEKK copolymers were successfully thermally classified, which provided a feasible means for the rapid thermal classification of PEKK copolymers and laid the foundation for subsequent research.
References
Garcia-Gonzalez D, Rodriguez-Millan M, Rusinek A, Arias A. Investigation of mechanical impact behavior of short carbon-fiber-reinforced PEEK composites. Compos Struct. 2015;133:1116–26. https://doi.org/10.1016/j.compstruct.2015.08.028.
Hassan EAM, Ge D, Yang L, Zhou J, Liu M, Yu M, Zhu S. Highly boosting the interlaminar shear strength of CF/PEEK composites via introduction of PEKK onto activated CF. Compos A Appl Sci Manuf. 2018;112:155–60. https://doi.org/10.1016/j.compositesa.2018.05.029.
Nassir N, Birch RS, Cantwell WJ, Wang QY, Liu LQ, Guan ZW. The perforation resistance of glass fibre reinforced PEKK composites. Polym Test. 2018;72:423–31. https://doi.org/10.1016/j.polymertesting.2018.11.007.
Perez-Martin H, Mackenzie P, Baidak A, Bradaigh CMO, Ray D. Crystallinity studies of PEKK and carbon fibre/PEKK composites: a review. Compos Part B Eng. 2021;223:109127. https://doi.org/10.1016/j.compositesb.2021.109127.
Zhu K, Tan H, Wang Y, Liu C, Ma X, Wang J, Xing H. Crystallization and mechanical properties of continuous carbon fiber reinforced polyether-ether-ketone composites. Fibers Polym. 2019;20:839–46. https://doi.org/10.1007/s12221-019-8791-5.
Wang W, Schultz JM, Hsiao BS. Dynamic study of crystallization- and melting-induced phase separation in PEEK/PEKK blends. Macromolecules. 1997;30:4544–50. https://doi.org/10.1021/ma970092l.
Cheng SZD, Ho RM, Hsiao BS, Gardner KH. Polymorphism and crystal structure identification in poly(aryl ether ketone ketone)s. Macromol Chem Phys. 1996;197:185–213. https://doi.org/10.1002/macp.1996.021970115.
Hsiao BS, Chang IY, Sauer BB. Isothermal crystallization kinetics of poly(ether ketone ketone) and its carbon-fiber-reinforced composites. Polymer. 1991;32:2799–805. https://doi.org/10.1016/0032-3861(91)90111-u.
Hallam B. Aromatic polyketones and preparation thereof, US Pat 3065205; 1962.
Colquhoun HM, Lewis DF. Synthesis of aromatic polyetherketones in trifluoromethanesulphonic acid. Polymer. 1988;29:1902–8. https://doi.org/10.1016/0032-3861(88)90410-7.
Iwakura Y, Uno K, Takiguchi T. Syntheses of aromatic polyketones and aromatic polyamide. J Polym Sci A-1-Polym Chem. 1968;6:3345–55. https://doi.org/10.1002/pol.1968.150061213.
Niume K, Toda F, Uno K, Iwakura Y. Catalytic Friedel-Crafts polyketone synthesis in PPA. J Polym Sci Polym Lett. 1977;15:283–6. https://doi.org/10.1002/pol.1977.130150505.
Ueda M, Oda M. Synthesis of aromatic poly(ether ketone)s in phosphorus pentoxide methanesulfonic-acid. Polym J. 1989;21:673–9. https://doi.org/10.1295/polymj.21.673.
Gay FP, Mary BC. Ordered polyetherketones, US Pat 4816556; 1989.
Smith KJ, Towle IDH, Moloney MG. Spherical, particulate poly(ether ketone ketone) by a Friedel Crafts dispersion polymerisation. RSC Adv. 2016;6:13809–19. https://doi.org/10.1039/c5ra25253a.
Gardner KH, Hsiao BS, Matheson RR, Wood BA. Structure, crystallization and morphology of poly (aryl ether ketone ketone). Polymer. 1992;33:2483–95. https://doi.org/10.1016/0032-3861(92)91128-o.
Hsiao BS, Gardner KH, Cheng SZD. Crystallization of poly(aryl ether ketone ketone) copolymers containing terephthalate/isophthalate moieties. J Polym Sci Part B Polym Phys. 1994;32:2585–94. https://doi.org/10.1002/polb.1994.090321604.
Gardner KH, Hisao BS, Faron KL. Polymorphism in poly(aryl ether ketone)s. Polymer. 1994;35:2290–5. https://doi.org/10.1016/0032-3861(94)90763-3.
Song P, Chen G, Wei Z, Zhang W, Liang J. Calorimetric analysis of the multiple melting behavior of melt-crystallized poly(l-lactic acid) with a low optical purity. J Therm Anal Calorim. 2013;111:1507–14. https://doi.org/10.1007/s10973-012-2502-4.
Di Lorenzo ML. Calorimetric analysis of the multiple melting behavior of poly(l-lactic acid). J Appl Polym Sci. 2006;100:3145–51. https://doi.org/10.1002/app.23136.
Yasuniwa M, Lura K, Dan Y. Melting behavior of poly(l-lactic acid): effects of crystallization temperature and time. Polymer. 2007;48:5398–407. https://doi.org/10.1016/j.polymer.2007.07.012.
Shieh Y-T, Liu G-L. Temperature-modulated differential scanning calorimetry studies on the origin of double melting peaks in isothermally melt-crystallized poly(l-lactic acid). J Polym Sci Part B Polym Phys. 2007;45:466–74. https://doi.org/10.1002/polb.21056.
Yasuniwa M, Tsubakihara S, Sugimoto Y, Nakafuku C. Thermal analysis of the double-melting behavior of poly(l-lactic acid). J Polym Sci Part B Polym Phys. 2004;42:25–32. https://doi.org/10.1002/polb.10674.
Kong Y, Hay JN. Multiple melting behaviour of poly(ethylene terephthalate). Polymer. 2003;44:623–33. https://doi.org/10.1016/s0032-3861(02)00814-5.
Wang ZG, Hsiao BS, Sauer BB, Kampert WG. The nature of secondary crystallization in poly(ethylene terephthalate). Polymer. 1999;40:4615–27. https://doi.org/10.1016/s0032-3861(99)00067-1.
Bassett DC, Olley RH, Alraheil IAM. On crystallization phenomena in PEEK. Polymer. 1988;29:1745–54. https://doi.org/10.1016/0032-3861(88)90386-2.
Lattimer MP, Hobbs JK, Hill MJ, Barham PJ. On the origin of the multiple endotherms in PEEK. Polymer. 1992;33:3971–3. https://doi.org/10.1016/0032-3861(92)90391-9.
Lee YC, Porter RS, Lin JS. On the double-melting behavior of poly(ether ether ketone). Macromolecules. 1989;22:1756–60. https://doi.org/10.1021/ma00194a043.
Tan SS, Su AH, Luo J, Zhou EL. Crystallization kinetics of poly(ether ether ketone) (PEEK) from its metastable melt. Polymer. 1999;40:1223–31. https://doi.org/10.1016/s0032-3861(98)00275-4.
Tardif X, Pignon B, Boyard N, Schmelzer JWP, Sobotka V, Delaunay D, Schick C. Experimental study of crystallization of polyetheretherketone (PEEK) over a large temperature range using a nano-calorimeter. Polym Test. 2014;36:10–9. https://doi.org/10.1016/j.polymertesting.2014.03.013.
Wei CL, Chen M, Yu FE. Temperature modulated DSC and DSC studies on the origin of double melting peaks in poly(ether ether ketone). Polymer. 2003;44:8185–93. https://doi.org/10.1016/j.polymer.2003.10.009.
Zhang ZY, Zeng HM. Effects of thermal-treatment on poly(ether ether ketone). Polymer. 1993;34:3648–52. https://doi.org/10.1016/0032-3861(93)90049-g.
Chelaghma SA, De Almeida O, Margueres P, Passieux J-C, Perie J-N, Vinet A, Reine B. Identification of isothermal crystallization kinetics of poly(ether-ketone-ketone) based on spherulite growth measurements and enthalpic data. Polym Crystal. 2020;3:e1014. https://doi.org/10.1002/pcr2.10141.
Choupin T, Fayolle B, Regnier G, Paris C, Cinquin J, Brule B. Isothermal crystallization kinetic modeling of poly(etherketoneketone) (PEKK) copolymer. Polymer. 2017;111:73–82. https://doi.org/10.1016/j.polymer.2017.01.033.
Choupin T, Fayolle B, Regnier G, Paris C, Cinquin J, Brule B. A more reliable DSC-based methodology to study crystallization kinetics: application to poly(ether ketone ketone) (PEKK) copolymers. Polymer. 2018;155:109–15. https://doi.org/10.1016/j.polymer.2018.08.060.
Choupin T, Fayolle B, Regnier G, Paris C, Cinquin J, Brule B. Macromolecular modifications of poly(etherketoneketone) (PEKK) copolymer at the melting state. Polym Degrad Stab. 2018;155:103–10. https://doi.org/10.1016/j.polymdegradstab.2018.07.005.
Roland S, Moghaddam M, Tence-Girault S, Fayolle B. Evolution of mechanical properties of aged poly(ether ketone ketone) explained by a microstructural approach. Polym Degrad Stab. 2021;183:109412. https://doi.org/10.1016/j.polymdegradstab.2020.109412.
Cortes LQ, Causse N, Dantras E, Lonjon A, Lacabanne C. Morphology and dynamical mechanical properties of poly ether ketone ketone (PEKK) with meta phenyl links. J Appl Polym Sci. 2016;133:43396. https://doi.org/10.1002/app.43396.
Coulson M, Cortes LQ, Dantras E, Lonjon A, Lacabanne C. Dynamic rheological behavior of poly(ether ketone ketone) from solid state to melt state. J Appl Polym Sci. 2018;135:46456. https://doi.org/10.1002/app.46456.
Arandia I, Mugica A, Zubitur M, Iturrospe A, Arbe A, Liu G, Wang D, Mincheva R, Dubois P, Mueller AJ. Application of SSA thermal fractionation and X-ray diffraction to elucidate comonomer inclusion or exclusion from the crystalline phases in poly(butylene succinate-ran-butylene azelate) random copolymers. J Polym Sci Part B Polym Phys. 2016;54:2346–58. https://doi.org/10.1002/polb.24146.
Arnal ML, Balsamo V, Ronca G, Sanchez A, Muller AJ, Canizales E, de Navarro CU. Applications of successive self-nucleation and annealing (SSA) to polymer characterization. J Therm Anal Calorim. 2000;59:451–70. https://doi.org/10.1023/a:1010137408023.
Cordova ME, Lorenzo AT, Mueller AJ, Hoskins JN, Grayson SM. A comparative study on the crystallization behavior of analogous linear and cyclic poly(epsilon-caprolactones). Macromolecules. 2011;44:1742–6. https://doi.org/10.1021/ma200394h.
Lorenzo AT, Arnal ML, Mueller AJ, Lin M-C, Chen H-L. SAXS/DSC analysis of the lamellar thickness distribution on a SSA thermally fractionated model polyethylene. Macromol Chem Phys. 2011;212:2009–16. https://doi.org/10.1002/macp.201100240.
Mary Michell R, Mueller AJ, Deshayes G, Dubois P. Effect of sequence distribution on the isothermal crystallization kinetics and successive self-nucleation and annealing (SSA) behavior of poly(epsilon-caprolactone-co-epsilon-caprolactam) copolymers. Eur Polym J. 2010;46:1334–44. https://doi.org/10.1016/j.eurpolymj.2010.03.013.
Mueller AJ, Lorenzo AT, Arnal ML. Recent advances and applications of “successive self-nucleation and annealing” (SSA) high speed thermal fractionation. Macromol Symp. 2009;277:207–14. https://doi.org/10.1002/masy.200950325.
Mueller AJ, Michell RM, Perez RA, Lorenzo AT. Successive self-nucleation and annealing (SSA): correct design of thermal protocol and applications. Eur Polym J. 2015;65:132–54. https://doi.org/10.1016/j.eurpolymj.2015.01.015.
Muller AJ, Arnal ML. Thermal fractionation of polymers. Prog Polym Sci. 2005;30:559–603. https://doi.org/10.1016/j.progpolymsci.2005.03.001.
Perez RA, Cordova ME, Lopez JV, Hoskins JN, Zhang B, Grayson SM, Mueller AJ. Nucleation, crystallization, Self-Nucleation and thermal fractionation of cyclic and linear poly(epsilon-caprolactone)s. React Funct Polym. 2014;80:71–82. https://doi.org/10.1016/j.reactfunctpolym.2013.10.013.
Pomatto ME, Moore RB. Crystallization kinetics and equilibrium melting temperature of poly (ether ketone ketone) with high terephthalate content utilizing fast scanning calorimetry. Polymer. 2023;271:125810. https://doi.org/10.1016/j.polymer.2023.125810.
Blundell DJ, Osborn BN. The morphology of poly(aryl-ether-ether-ketone). Polymer. 1983;24:953–8. https://doi.org/10.1016/0032-3861(83)90144-1.
Jonas A, Legras R. Thermal-stability and crystallization of poly(aryl ether ether ketone). Polymer. 1991;32:2691–706. https://doi.org/10.1016/0032-3861(91)90095-z.
Hoffman JD, Weeks JJ. Melting process and equilibrium melting temperature of polychlorotrifluoroethylene. Res Natl Bur stand. 1962;66:13–28. https://doi.org/10.6028/jres.066A.003.
Ho RM, Cheng SZD, Hsiao BS, Gardner KH. Crystal morphology and phase identifications in poly(aryl ether ketone)s and their copolymers. 1. Polymorphism in PEKK. Macromolecules. 1994;27:2136–40. https://doi.org/10.1021/ma00086a023.
Chen H, Cebe P. Investigation of the rigid amorphous fraction in Nylon-6. J Therm Anal Calorim. 2007;89:417–25. https://doi.org/10.1007/s10973-007-8215-4.
Chen H, Cebe P. Vitrification and devitrification of rigid amorphous fraction of PET during quasi-isothermal cooling and heating. Macromolecules. 2009;42:288–92. https://doi.org/10.1021/ma802104a.
Righetti MC, Tombari E. Crystalline, mobile amorphous and rigid amorphous fractions in poly(l-lactic acid) by TMDSC. Thermochim Acta. 2011;522:118–27. https://doi.org/10.1016/j.tca.2010.12.024.
Righetti MC, Tombari E, Angiuli M, Di Lorenzo ML. Enthalpy-based determination of crystalline, mobile amorphous and rigid amorphous fractions in semicrystalline polymers—poly(ethylene terephthalate). Thermochim Acta. 2007;462:15–24. https://doi.org/10.1016/j.tca.2007.06.003.
Song M, Hourston DJ. Temperature-modulated differential scanning calorimetry—IX. Some comments on the rigid amorphous fraction in semi-crystalline poly(ethylene terephthalate). J Therm Anal Calorim. 1998;54:651–7. https://doi.org/10.1023/a:1010163113898.
Wei Z, Song P, Zhou C, Chen G, Chang Y, Li J, Zhang W, Liang J. Insight into the annealing peak and microstructural changes of poly(l-lactic acid) by annealing at elevated temperatures. Polymer. 2013;54:3377–84. https://doi.org/10.1016/j.polymer.2013.04.027.
Xu H, Cebe P. Heat capacity study of isotactic polystyrene: dual reversible crystal melting and relaxation of rigid amorphous fraction. Macromolecules. 2004;37:2797–806. https://doi.org/10.1021/ma035961n.
Author information
Authors and Affiliations
Contributions
Conceptualization was contributed by JS and PS; methodology was contributed by ZW; formal analysis and investigation were contributed by JS and PS; writing—original draft preparation was contributed by JS; writing—review and editing, was contributed by PS, ZW; resources were contributed by GL and QS; Supervision was contributed by ZW and YL.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Shang, J., Song, P., Li, G. et al. Crystallization and melting behavior of poly(ether ketone ketone) (PEKK) copolymers synthesized by facile one-step method. J Therm Anal Calorim 148, 11727–11741 (2023). https://doi.org/10.1007/s10973-023-12518-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-023-12518-1