
Abstract
The development of new materials called geopolymers is described, which at the turn of the nineties brought a new state of the art in material design through the so-called wet process resulting in a specific amorphous state. The classical configuration of glasses prepared by quenching is used for a joint appraisal and judgment. We can use the comparison and description of the known form of organic polymers with the so-called mers-structure. The formation involves a sol–gel polycondensation chemical reaction also known in the case of organic polymers. The formation is described using aluminosilicate oxide in IV-fold coordination with alkaline polysilicates to form polymeric Si–O–Al chains through amorphous to semi- and hypo-crystalline three-dimensional silico-aluminate structures. The revision of structural units and their interconnection is evaluated, and it turns out that the common factor of the multiparty description is the existence of bridging and non-bridging oxygen. The review provides a detailed overview of opinion while reminding that the historical origin of the field falls within the purview of the JTAC journal.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
It is important to recall that the thermoanalytical pages of the Journal of Thermal Analysis and Calorimetry (JTAC) dared to publish two then unnamed material-oriented papers, thus initiating a new realm of materials called geopolymers [1] and novel applications leading to the description of bridged oxygen in the field of silica-based glasses [2] and inorganically clarified bioactivities. These two pioneer articles, which become afterword intertwined, emanated together conceptually aiming to the later understanding the role of bridged oxygen in non-crystalline materials. Certainly it became associated with various support ideas such as early polysialate studies [3,4,5,6] and premature revisions on silica ionic concepts [7,8,9,10] until then rather unfamiliar. Therefore, it is important to realize that it was the de facto courage of the JTAC editorial office to dare to publish an unaccredited topic. Moreover, it was completely new and did not immediately contribute to the journal's reputation. In turn, however, it brought desired fame and today's profit impact factor to the originally unknown periodical exceeding four.
Since then, of course, much has changed, the mechanisms of origin and descriptions of structures have been specified in a number of hypotheses of both polysialates [11,12,13,14,15,16,17] and ionic systems [18,19,20,21,22,23], as evidenced by the publication of hundreds of works, especially review papers on geopolymers [24,25,26,27,28,29,30,31,32,33,34,35]. Subsequent works then pointed out the interconnectedness of these topics [36, 37]. Despite the fact that this is a popular topic, it seems that it would be good to recall some aspects of the development and to clarify and link the existing design views on the structure of these materials. This is especially evident from the comparison of oxide glass structures prepared by melt cooling versus amorphous polysialates prepared by cold chemical reactions. In terms of structural units and bonds, the two variants thus prepared seem to have much in common, as shown in next paragraph.
Glassy versus amorphous states
Non-crystalline substances are usually named glassy and/or amorphous. The term glassy or vitreous derives from the traditional preparation of silicate glasses for practical purposes, when for the standard temperature regime of natural cooling, a so-called batch composition was sought that would meet the given requirement. Successive development has shown that the glassy state can be prepared for most materials if a suitable method of their rapid cooling (quenching) is established [38,39,40,41], which also led to the preparation of unexpected metallic glasses [42,43,44]. Thus, a number of publications have appeared describing various methods and processes of glass preparation, particularly specifying cooling techniques, where the melt is usually subjected to intensive heat removal in contact with the cooling surface of the cold gadget, causing unwanted crystallization, i.e., equilibrium phase transformation. The emergence of the glassy state is linked to the so-called glass transformation, from the description of which the specific criteria of glass stability and the ability of melts to form a glassy state are derived [45, 46]. Glass formation is therefore limited to the phase range between the melt and the solid phase, which is determined by the speed (via method) of cooling, i.e., sufficient heat removal. This also led to the preparation of unorthodox oxide glasses and the use of special quenching techniques [22, 47, 48] which later also led to the clarification of the function of non-bridging oxygen [22, 47, 48].
For illustration, Fig. 1, left column, displays an example of melting configuration and is usually described via the so-called T–T–T (or better C–T) diagrams [49,50,51] showing the possibilities of achieving the glassy state for a given cooling method (cooling rate ϕ) given by types of mechanical quenching (see schematic method markers). It is obvious that the formation of such a glassy state is a matter of the heated conditions taking place on the cooled surface, preferably within the layer of the entirely solidified melt.

Comparison of two preparation routes. Left: glass-formation via quenching using various rapid cooling techniques where glass formation is given by the ability to remove heat from the solidifying interface quickly enough without crystallization occurring. Usually schematized using T-T-T diagrams is seemly a model of metallic glasses, and glass-formation ability is related to the value of associated glass transition Tg. Particularly, it shares decryption for glasses of brand PdCuSi, PdSi, AuGe,SI or even Ni (Tg > 1/4) while difficult vitrifying oxide mixture is (Fe,Mn)2O3–(Bi,B)2O3. (Tg ≈ 2/3). Right: Procedure of the process route for the preparation of amorphous polysialates by chemical reactions in the presence of a liquid state creating a solid non-crystalline substance comparable with glasses
By default, the structure of pure vitrified SiO2 can generally be considered as a continuous not unshared (i.e., non-bridging). Tetrahedra with n-type bridging O are referred to as specific species Qn (n = 1, 2, 3, 4). Initial configurations can be constructed such that Si atoms are introduced random network of corner-sharing SiO4 tetrahedra where each oxygen bridges two tetrahedra. It relates to the ratio between the molar amounts of oxygen and silicon atoms R = n(O)/n(Si). The structure can then be characterized by the amalgamation of the so-called Q units traditionally derived on the basis of the above ratio, namely from Q0 to Q4 following progression \(Q0=\text{SiO}_{4}^{4-}, Q1=\text{Si}_{2}\text{O}^{6-}_{7}, Q2= \text{SiO}_{3}^{2-}, Q3=\text{Si}_{2}\text{O}^{2-}_{5} \) and Q4 being SiO2. A series of equations can then be derived as \(2\text{SiO}_{4}^{4-}=\text {Si}_{2}\text{O}_{7}^{6-}+\text{O}^2\) and/or \(6\text{SiO}_{3}^{2-}=2\text{Si}_{2}\text{O}_{5}^{2-}+2\text{SiO}_{4}^{4-}\).
When a modifier such as an alkali metal oxide is added, the additional oxygen added causes some network breakdown. Although each Si remains four times coordinated with O, nevertheless some of the positions become resonant. In the first of their groupings, the O's are randomly with n-fold Si–Si coordination constraints, given by equivalent proportions of Qn species usually determined from NMR. For example, in the case of (K2O)0.2(SiO2)0.8, the Q4:Q3:Q2 ratios are 0.57:0.35:0.08, and the coordination thus achieved is almost perfect. If O atoms are added to the center of each Si–Si bond the individual O atoms are then assigned close to Si atoms, which now have only two or three neighbors (double or triple Si–Si coordination). In the ideal modified random network model, the ratio of non-bridging and bridging oxygens is given by the relation 2y/(2 + y), where y is bound concentrationally as x/(1 − x). Alkali atoms are usually added randomly the Si–O coordination constraint remains respected.
On the other hand (the right column) the preparation of amorphous geopolymers discussed above is realized by chemical processing in bulk creating thus a solid substance, via so-called wet treating, and shown by successive steps in Fig. 1. It is obvious that this is a different way of creating a non-crystalline state usually referred to as amorphous [11, 25, 30,31,32,33,34,35]. This was also reflected in the search for a more recent structural description, which was already presented 40 years ago by J. Davidovits himself [1, 4]. The concept of geopolymers was created and described as a family of isomorphous compounds with a zeolitic three-dimensional crystal structure, while the following research was focused more on the application area. Davidovits [1, 4] established a coherent scientific terminology based on different chemical units, essentially for silicate and aluminosilicate materials, classified according to the atomic ratio Si:Al: Si:Al = 0 (silox), Si:Al = 1 (sialate), Si:Al = 2 (sialate-siloxo), Si:Al = 3 (sialate-disiloxo) and Si:Al > 3 (sialate) [6]. Conceptually, this is not much different from above readings on silica glasses showing a group connectivity and related specification of so-called Q-motives [19, 52,53,54].
Structural interconnection of geopolymers and their unified understanding
By default, the very structure of pure vitrified SiO2 can generally be considered as a continuous random network of corner-sharing SiO4 tetrahedra where each oxygen bridges two tetrahedra. When a modifier such as an alkali metal oxide is added, the additional oxygen added causes some network breakdown. Although each Si remains four times coordinated with O, some of the positions become resonant. In the first of their groupings, the O's are not unshared (i.e., non-bridging). Tetrahedra with n-type bridging O are referred to as specific species Qn. Initial configurations can be constructed such that Si atoms are introduced randomly with n-fold Si–Si coordination constraints, given by equivalent proportions of Qn species usually determined from NMR. For example, in the case of (K2O)0.2(SiO2)0.8, the Q4:Q3:Q2 ratios are 0.57:0.35:0.08, and the coordination thus achieved is almost perfect. O atoms are added to the center of each Si–Si bond, and individual O atoms are then assigned close to Si atoms, which now have only two or three neighbors (double or triple Si–Si coordination). In the ideal modified random network model, the ratio of non-bridging and bridging oxygens is given by the relation 2y/(2 + y), where y is bound centrally as x/(1 − x). Alkali atoms are usually added randomly respecting the Si–O coordination constraint.
Such related aluminosilicate gels in the form of zeolite precursors are mostly synthesized in a composition characterized by the general formula Mm[–(Si–O)z–Al–O]n∙wH2O, where Mm are modifying cations (mostly Na, K, Ca, Mg) and where m is the degree of polycondensation while z means structural classification (usually 1, 2, 3, …). Configurationally, the tetrahedra of SiO4 and AlO4 are bound to each other by oxygen bridges, which thus form both chains and rings based on Si–O–Al. The positively charged Mm ions must be compensated by the negative charge of the four-coordinated Al. The gel-like structure created is partially amorphous or semi- or nano-crystalline, depending on both the amount of starting solid matter and its nature (character of the raw materials), as well as the conditions of the wet reaction (pH). The amorphous state becomes more favorable for a higher concentration of solid precursor in the preparation suspension. XRD patterns of commercial molten glass and wet-synthesized inorganic polymer were compared [55] showing broad peaks typical of amorphous phases, which, however, are slightly shifted to higher angle values θ for the inorganic polymer, which questions the opinion of a relatively denser structure than that of the analogous commercial glass. A deeper deconvolution of such broad peaks in the XRD patterns can also allow a quantitative estimation of the degree of non-crystallinity, which we can estimate as the gradation of amorphicity.
In practice, the reacting gel subsequently hardens into solid geopolymers resembling to a certain extent the quenched-formation of glasses during solidification of the melt (cf. Fig. 1.). This can be characterized in a number of ways somehow similar to classical glasses. For example, it can be a description in terms of the introduction of the main components (alumina and silica), their structure (Al–O and Si–O tetrahedral units in a random 3D framework), the balancing role of alkali ions, etc. The specific circumstances of low-temperature synthesis, such as the condensation temperature of aluminum oxide and silica sources at high pH and the alteration of different types of water glasses (their rich structure and thus adjustability), are worth the further clarification process usually leading to different degrees of achieved amorphicity. A mutual comparison of the possible structures of constrained (melt-frozen) glasses and wet-casted amorphous geopolymers is shown in Fig. 2 with reference to the role of bonding oxygen.

Left: explanatory structure of silica glass with indicated important sites and bonds; middle: the typical aluminosilicate melt-quenched glass and right: its amorphous version prepared by geopolymeric wet-cast
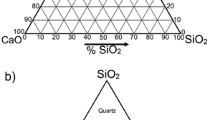
Some formalism has been developed to investigate the introduced structural units, mostly in the sense of fragments such as [–Si–O–Al–O–] called sialate units or even polysialates when they are simultaneously condensed. Other suggested measures can be different Si:Al ratios because geopolymerization involves a chemical reaction of aluminosilicate oxides, especially Al3+ in IV-fold coordination to alkaline polysilicates enabling the formation of polymeric Si–O–Al bonds. Structurally three-dimensional, amorphous to semi-crystalline silico-aluminate structures are of the poly(sialate) type (–SiO–Al–O–), the poly(sialate-siloxo) type (–Si–O–Al–O–Si–O–), type Poly(sialate-disiloxo) (–Si–O–Al–O–Si–O–Si–O–). The atomic ratio Si:Al is considered in the most common integer form (1, 2 and 3), but even non-integer ratios between 1:1 and 1:3 can be assumed as variable combinations of basic units, provided that the content of charge-balancing cations is suitable and often controlled by the water content. Units with a value of Si:Al > 3 are then referred to as sialate and polysialate geopolymers. In terms of the majority composition of the Earth's crust, which consists of siloxo-sialates and sialates (see Fig. 3), the common series of feldspars albite-anorthite (NaAlSi3O8–CaAl2Si2O8) can be described as poly (sialate-disiloxo) for albite to poly(disialate) for anorthite.

Mechanisms of geopolymers formation [56]. Reprinted from Ion Exchange in Geopolymers, J. R. Gasca-Tirado, A. Manzano-Ramírez, E. M. Rivera Muñoz, R. Velázquez-Castillo, M. Apátiga-Castro, R. Nava and A. Rodríguez-López, In: New Trends in Ion Exchange Studies (S. Karakus, editor), London: IntechOpen, 2018. Licensed under CC BY 3.0, https://creativecommons.org/licenses/by/3.0/
Theory of hypo-crystalline materials and their ‘mers’ framework
It seems that so far not enough attention has been paid to the basic structural disposition, although we know sufficiently detailed studies on the reaction mechanism, reorganization and hardening. It seems at first glance that the inherent stoichiometry can be understood analogously to both organic and inorganic –mers, well known in classical polymer chains. Such a generalized sphere of description can be covered by the term so-called hypo-crystalline materials (as a newly introduced term more suitable for the corresponding terminology), which is specific in that it includes regular AOn polyhedra such as SiO4, AlO3, AlO4, BO4, BO3, PO4. The connecting function of the bridges formed by all n-atoms of oxygen appears to be important. If oxygen is bridged by two central cations, A- (e.g., (SiO4/2) as AOn/2), then the oxygen atoms are also bound to the A-cation by a double bond (so-called terminal bonds, known from single-component glass composed of, e.g., from phosphorus oxides containing –mers (O=P(–O–)3). The most suitable coordination formula takes the form (AOi/1O(n−i)/2), where (PO1/1O3/2) can serve as an illustrative example of a coincidence manifold stoichiometry. This is consistent with the analogy of organic polymers in terms of (AOi/1O(n−i)/2), which can be considered as (n − i)-functional –mer. Aluminosilicates composed of tetrahedral alumina and silica units contain mer-units AOn/2, which upon condensation become relocatable due to their long lifetimes. This become comparable to the degree of immobility in the high-temperature state of melts/glasses. Similar to oxide glasses, the randomly interconnected network is continuous (random network). A complex copolymer system (containing complements of moderators electropositive elements typically alkali oxides, M2O or other metal oxides) persists due to the function of modifying oxides. In such systems (melts/glasses/macromolecules), additions to a single-component (tetragonally cross-linked) solution result in the breakdown of A–O–A bridging bonds and the formation of so-called non-bridging oxygens. They can be described by a set of equations:
and
which are actually responsible for the dissociation concerning one molecule of the modifying oxide and the subsequent decay bridging bonds (A–O–A). In the case of typical modifying oxides, the equilibrium tends to shift toward the products, and the arrangement is readily inferred from the initial melt/glass/polymer stoichiometry.
The mineralogical composition of the studied geopolymers was variously identified as, for example, for the dependence of the bending strength on the Al/(Na½Ca) ratio, which can be calculated using the derived formulas nM2O∙pCaO∙Al2O3∙xSiO2∙yH2O. Unlike the Si/Al ratio, which was not a significant factor in the range of 1.5–2.1; however, a maximum flexural strength of 10.2 MPa was achieved for material with Al/(Na½Ca)¼ where XRD analysis identified both quartz (SiO2), albite (NaAlSi3O8) and muscovite (KAl2(AlSi3O10)(OH)2) as usually dominant compounds in most mixtures. Diopside (CaMgSi2O6), akermanite (Ca2MgSi2O7), hematite (Fe2O3), illite (K0.65Al2(Al0.65Si3.35O10)(OH)2), orthoclase (KAlSi3O8) and microcline (KAlSi3O8) were found in lower amounts. The main crystalline phases identifiable in the engineered geopolymers were identical to those found in the raw precursor, indicating only partial geopolymerization as well as the presence of significant amounts of unreacted particles. The contents of quartz, hematite and orthoclase gradually decrease with increasing curing reaction temperature. For geopolymers cured around 60 °C, the formation of zeolitic phases was observed, the total amount of which then further increased at 80 °C curing, even though the identified phases did not soften. On the other hand, the silicate module of the alkaline activator, SM, significantly influences the type of zeolitic phase. Sodalite (Na8(Al6Si6O24)Cl2) was found only in mixtures with SM = 0.8, i.e., with the highest dose of sodium hydroxide. Geopolymers with SM = 1.0, 1.2 and 1.4 contained gismondine (CaAl2Si2O8·4H2O) and geopolymers with SM = 1.0 also contained chabazite (Ca,Na2,K2,Mg)Al2Si4O12·6H2O). Diopside (CaMgSi2O6), akermanite (Ca2MgSi2O7), hematite (Fe2O3), illite (K0.65Al2(Al0.65Si3.35O10)(OH)2), orthoclase (KAlSi3O8) and microcline (KAlSi3O8) were found in lower amounts. The detailed nature of the gradual adjustment of the composition is still a subject of research, as is the possibility of inserting modifying admixtures from organic to purely organic (asphalts).
Bridging and non-bridging oxygen and its plausible impact to bioactivity
The nature of the oxygen bond, the correlation between the structural parameters characterized by the anionic constitution [2, 57, 58] is also interesting for the definition of bioactivity. A simple way to describe the solidified composition of the melt/glass is to use the so-called Steevel parameters X and Y [2, 59], which indicate the average number of non-bridging oxygen ions (NBO) and bridging oxygen ions (BO) per polyhedron in the glass lattice. They can be calculated using the molar composition of the glass assuming the following equations, X = 2R − Z and Y = 2Z − 2R, where Z and R denote the mean number of all types of oxygen per polyhedron and the ratio of the total number of oxygens to the total number of glass-forming cations in the glass, respectively. Regarding the classical sodium-calcium-silica system, pure silica glass SiO2, pseudobinary Na2O–2SiO2 glass, and pseudoternary CaO–Na2O–2SiO2 glass are characterized by (X, Y) = (0, 4); (1, 3) and (2, 2), respectively. When X > 2 and Y < 2, the concentration of free ions (FO) is noticeably greater, and glasses in this range of composition are commonly called invert. For bioactive glass–ceramic systems of the Na2O–CaO(MgO)–SiO2(–P2O5) type, the Y parameter was correlated with the bioactivity assessed by the in vitro cross-linking test after immersion in simulated body fluids (SBF) [21]. It was concluded that Y = 2 in the residual glass in the glass–ceramic phase is a favorable condition for higher biological activity, while Y > 3 reduces biological activity. Focusing on the structural parameter X related to the NBO value, its correlation with bioactivity in the Na2O–CaO–SiO2–P2O5 system is enabled by evaluating the bioactivity index IB (empirically introduced by Hench [60]). A linear correlation between X and IB with a positive slope was found in the range 1.5 < X < 2, while IB = 0 for X < 1.5. Accordingly, considering the composition of BO and NBO expressed by the Steevel parameters X and Y, the region of bioactive composition can be suggested as 1.5 < X < 2 and 2 < Y < 2.5. Assuming that silicate ions are presented as linear and branching chains according to the general formula [SixO3x+1](2x+2)−, then the compositional dependence of the relative proportion of NBO in the binary glass-forming melts Na2O–SiO2 and CaO–SiO2 can be estimated [2, 9, 61] and silicate ions in the equilibrium state of polycondensation reactions are described by the following equation:
The inherent equilibrium constant kxy of this equation can be approximated using that for the lowest k-members k11, i.e., x = 1 and y = 1. In view of the constitutions of BO, NBO and FO ions, the equation O° + O2− ⇄ 2O− is apparently expressed as the inverse reaction.
where a can be calculated by the Temkin’s equation [10] as is equivalent to the ion fraction. More details are available in associated papers [2, 21, 36, 37] and falling thus beyond this review.
In conclusion, it can be said that the type of binding oxygen is essential in all types of silicate structures, both quenched glasses and wet-cast geopolymers, which is often forgotten. Especially known is its role in the biological groups that may be developmentally responsible for the emergence of living structures [62], bioactivity of glasses, where the formation of the necessary bone apatite depends on the relative proportion of NBO in the glass. For example, it is the currently analyzed type CaO–Na2O–SiO2 and in the glass–ceramic alternative, regardless of whether P2O5 was added. This points to a close connection between the structural properties of the glass characterized by the activity of oxygen ions and the surface chemical processes of bone apatite formation not included further. The bioactivity index for an active implant then depends both on the rate of formation of the calcium phosphate film and the time of crystallization to hydroxyl-carbonate apatite, and on the selective absorption of extracellular proteins on the growing hydroxyapatite layer, which control cell attachment, differentiation and growth. The defective structure of the growing hydroxyapatite crystals is probably responsible for the preferential adsorption of growth factors. Recent theories point to metastable hydrated silica molecules, such as penta-coordinated Si(OH)5 ions, which can increase the crystallization potential on surfaces and provide specific binding motivation for the activation of biological groups [62,63,64]. The coexisting three-membered silicon bonds are energetically metastable, and the Si–O–Si bridging bonds then allow, for example, the –COOOH radical or amino acids to interact with the trisiloxane ring. COOH thus polarizes the Si–O–Si bond and opens it into a chain where, like the five-level Si–OH complex, it acts as an inorganic enzyme, which provides a favorable pathway for polypeptide synthesis. This may explain the often mentioned biological activity of clays.
Discussion and conclusions notes
Geomaterials [65,66,67,68] have not only become a representative of modern material with a future, but they have also become a suitable focus of applications of the methods of thermal science and analysis [16, 23, 36, 46, 49, 51, 54]. Involved procedures suited as a useful implement in studying polysialates thermal relations where the involved heat impact always plays a significant role as an explicit tool in two interconnected forms [51]:
-
1.
as an industrial power applied for working geopolymeric and other glassy materials with desired properties by means of controlled heat input and/or removal,
-
2.
equally as an instrumental agent for modern analysis which, however, is a destructive technique studying the relationship between material’s properties and its temperature response while the sample is heated and/or cooled in a controlled manner when heat acts as its own reagent.
Both connotations are a significant part of the up-to-date material aimed thermodynamics needing an understanding of constrains and other intricacies of glassiness and amorphicity [51] in terms of their dynamic preparation and subsequent study [36, 45, 51, 69]. However, the detailed examination of related material properties is in many modern books not treated enough properly when the interpretation of thermodynamics (as classical textbook ‘thermostatics’) is not solved with the necessary emphasis on the needs of the solid-state thermal treatment [70,71,72]. The same applies to the recent approach toward new books on thermal analysis [72, 73]. Even in the field of geomaterials [65,66,67,68] a more modern approach to dynamic share of thermodynamics involving non-stoichiometry or glass formation should be applied [51], including its truly dynamic nature [41, 51, 69], where preparation and analysis take place in real off-equilibrium conditions, and the validity of thermodynamic lessons must thus be appropriately adapted [37, 51, 69].
References
Davidovits J. Geopolymers. J Therm Anal. 1991;37(8):1633–56.
Koga N, Strnad Z, Sestak J, Strnad J. Thermodynamics of non-bridging oxygen in silica bio-compatible glass-ceramics—mimetic material for the bone tissue substitution. J Therm Anal Calorim. 2003;71(3):927–37.
Davidovits J, Davidovics M. Geopolymer: room-temperature ceramic matrix for composites. In: Proceedings of the 12th annual conference on composites and advanced ceramic materials: ceramic engineering and science proceedings. 1988, pp 835–41
Davidovits J. Geopolymers and geopolymeric materials. J Therm Anal. 1989;35(2):429–41.
Barrer RM. Some researched on silicates: mineral synthesis and metamorphosis. Trans Br Ceram Soc. 1957;56:155–61.
Gluchovskij VD. Gruntosilikaty. Kiev: Gosstrojizdat; 1959.
Knight CTG, Balec RJ, Kinrade SD. The structure of silicate anions in aqueous alkaline solutions. Angew Chem Int Ed. 2007;46(43):8148–52.
Toop GW, Somis CS. Some new ionic concepts of silicate slags. Can Metall Q. 1962;1(2):129–52.
Masson CR. Anionic constitution of glass-forming melts. J Non-Cryst Solids. 1977;25(1):1–41.
Těmkin M. Mixtures of fused salts as ionic solutions. Acta Physicoch URSS. 1945;20:4511–9.
Davidovits J. Geopolymers: ceramic-like inorganic polymers. J Ceram Sci Technol. 2017;8(3):335–50.
Kriven WM, Bell JL, Gordon M. Microstructure and microchemistry of fully-reacted geopolymers and geopolymer matrix composites. In: International symposium on recent advances in ceramic matrix composites. Nashville, 2003, pp 227–50.
Rahier H, VanMele B, Biesemans M, Wastiels J, Wu X. Low-temperature synthesized aluminosilicate glasses. 1. Low-temperature reaction stoichiometry and structure of a model compound. J Mater Sci. 1996;31(1):71–9.
Rowles MR, O’Connor BH. Chemical and structural microanalysis of aluminosilicate geopolymers synthesized by sodium silicate activation of metakaolinite. J Am Ceram Soc. 2009;92(10):2354–61.
Kriven WM. Inorganic polysialates or “geopolymers.” Am Ceram Soc Bull. 2010;89(4):31–4.
Šestak J, Foller B. Some aspects of composite inorganic polysialates. J Therm Anal Calorim. 2012;108(2):511–7.
Provis JL, Palomo A, Shi CJ. Advances in understanding alkali-activated materials. Cem Concr Res. 2015;78:110–25.
Mysen BO, Shang J. Evidence from olivine/melt element partitioning that nonbridging oxygen in silicate melts are not equivalent. Geochim Cosmochim Acta. 2005;69(11):2861–75.
Machacek J, Gedeon O, Liska M. Group connectivity in binary silicate glasses. J Non-Cryst Solids. 2006;352(21–22):2173–9.
Greaves GN, Sen S. Inorganic glasses, glass-forming liquids and amorphizing solids. Adv Phys. 2007;56(1):1–166.
Šestak J, Strnad Z, Strnad J, Holecek M, Koga N. Biomedical thermodynamics and implantology aspects of biocompatible glass-ceramics and otherwise modified inorganic materials and surfaces. In: 9th Conference of the European-society-of-glass-science-and-technology/annual meeting of the international-commission-on-glass. Trencin, SLOVAKIA; 2008, pp 329.
Závěta K, Šesták J. Structure, bridging oxygen and magnetic properties of Fe-rich oxide glasses. In: 21st International congress on glass. Prague: Dům techniky ČVTS; 1977, pp 399.
Šesták J. Nonbridging oxygen in silica biocompatible glass ceramics and magnetic properties of Fe2O3 added borate glasses. In: Šesták J, Holeček M, Málek J, editors. Some thermodynamic, structural and behavioral aspects of glassy and amorphous materials. Plzeň: OPS-ZČU Plzeň; 2009. p. 128–51.
Komnitsas K, Zaharaki D. Geopolymerisation: a review and prospects for the minerals industry. Min Eng. 2007;20(14):1261–77.
Khale D, Chaudhary R. Mechanism of geopolymerization and factors influencing its development: a review. J Mater Sci. 2007;42(3):729–46.
Duxson P, Fernández-Jiménez A, Provis JL, Lukey GC, Palomo A, van Deventer JSJ. Geopolymer technology: the current state of the art. J Mater Sci. 2007;42(9):2917–33.
Li C, Sun HH, Li LT. A review: the comparison between alkali-activated slag (Si plus Ca) and metakaolin (Si plus Al) cements. Cem Concr Res. 2010;40(9):1341–9.
Lopes AC, Martins P, Lanceros-Mendez S. Aluminosilicate and aluminosilicate based polymer composites: present status, applications and future trends. Prog Surf Sci. 2014;89(3–4):239–77.
Palomo A, Krivenko P, Garcia-Lodeiro I, Kavalerova E, Maltseva O, Fernandez-Jimeneza A. A review on alkaline activation: new analytical perspectives. Mater Constr. 2014;64(315):e022.
Nikolov A, Nugteren H, Rostovsky I. Optimization of geopolymers based on natural zeolite clinoptilolite by calcination and use of aluminate activators. Constr Build Mater. 2020;243:118257.
Cong PL, Cheng YQ. Advances in geopolymer materials: a comprehensive review. J Traffic Transp Eng Engl Ed. 2021;8(3):283–314.
Castillo H, Collado H, Droguett T, Vesely M, Garrido P, Palma S. State of the art of geopolymers: a review. E-Polymers. 2022;22(1):108–24.
Zribi M, Baklouti S. Phosphate-based geopolymers: a critical review. Polym Bull. 2022;79(9):6827–55.
Longhi MA, Rodriguez ED, Walkley B, Eckhard D, Zhang ZH, Provis JL, et al. Metakaolin-based geopolymers: efflorescence and its effect on microstructure and mechanical properties. Ceram Int. 2022;48(2):2212–29.
Marvila MT, Azevedo ARG, Vieira CMF. Reaction mechanisms of alkali-activated materials, a review. Rev IBRACON Estrut Mater. 2021;14(3):e14309.
Šesták J, Koga N, Šimon P, Foller B, Roubíček P, Wu N-LN. Amorphous inorganic polysialates: geopolymeric composites and the bioactivity of hydroxyl groups. In: Šesták J, Šimon P, editors. Thermal analysis of micro, nano- and non-crystalline materials: transformation, crystallization, kinetics and thermodynamics. Dordrecht: Springer; 2013. p. 441–60.
Šestak J. Non-bridging oxygen in silica biocompatible glasses, ceramics, polysialates and geopolymers. In: Sestak J, editor. Thermal analysis and thermodynamic properties of solids. 2nd ed. Amsterdam: Elsevier; 2021. p. 437–58.
Šestak J. Miracle of reinforced states of matter. Glasses: ancient and innovative materials for the third millennium. J Therm Anal Calorim. 2000;61(1):305–23.
Collins LE. Overview of rapid solidification technology. Can Metallu Q. 1986;25(1):59–72.
Kramer MJ, Mecco H, Dennis KW, Vargonova E, McCallum RW, Napolitano RE. Rapid solidification and metallic glass formation—experimental and theoretical limits. J Non-Cryst Solids. 2007;353(32–40):3633–9.
Šesták J. The art and horizont of non-equilibriated and disordered states and the new glassy phase rapid formation techniques. Glass Sci Technol. 1997;70(1):153–63.
Kawazoe Y, Carow-Watamura U, Louzguine DV. A brief introduction to bulk metallic glasses: datasheet from condensed matter. In: Carow-Watamura U, editor. Phase diagrams and physical properties of nonequilibrium alloys, vol. 37C3. Berlin: Springer; 2013. https://doi.org/10.1007/978-3-662-57920-6_1.
Illeková E, Šesták J. Crystallization of metallic micro-, nano-, and non-crystalline alloys. In: Šesták J, Šimon P, editors. Thermal analysis of micro, nano- and non-crystalline materials: transformation, crystallization, kinetics and thermodynamics. Dordrecht: Springer; 2013. p. 257–89.
Prasad N. Introduction to metallic glasses. Metallic glass–based nanocomposites. Boca Raton: CRC Press; 2019.
Queiroz CA, Šestak J. Aspects of the non-crystalline state. Phys Chem Glass Eur J Glass Sci Technol Part B. 2010;51(3):165–72.
Kozmidis-Petrovic A, Šestak J. Forty years of the Hruby glass-forming coefficient via DTA when comparing other criteria in relation to the glass stability and vitrification ability. J Therm Anal Calorim. 2012;110(2):997–1004.
Šestak J, Šestakova V, Třiska A, Zavěta K. Glass-formation, phase-relations and magnetic-properties of the splat-quenched system of laser-melted (FE, MN)2O3-(BI, B)2O3. J Therm Anal Calorim. 1988;33(3):789–95.
Iordanova R, Dimitriev Y, Dimitrov V, Kassabov S, Klissurski D. Glass formation and structure in the system MoO3–Bi2O3–Fe2O3. J Non-Cryst Solids. 1998;231(3):227–33.
Šestak J. Use of phenomenological kinetics and the enthalpy versus temperature diagram (and its derivative—DTA) for a better understanding of transition processes in glasses. Thermochim Acta. 1996;280:175–90.
Schumacher O, Marvel CJ, Kelly MN, Cantwell PR, Vinci RP, Rickman JM, et al. Complexion time-temperature-transformation (TTT) diagrams: opportunities and challenges. Curr Opin Solid State Mater Sci. 2016;20(5):316–23.
Šesták J. Thermal analysis and thermodynamic properties of solids. 2nd ed. Amsterdam: Elsevier; 2021.
Machaček J, Gedeon O. Q-species in alkali-disilicate glasses. Ceram Silik. 2003;47(2):45–9.
Liška M, Klement R, Machaček J, Gedeon O. Inverse thermodynamic modelling of glass from Raman spectroscopical and molecular dynamics results. Phys Chem Glasses. 2005;46(2):108–11.
Liška M, Macháček J, Gedeon O. Molecular dynamics of the Na2O–MgO–SiO2 system. Glass Sci Technol. 2004;77(1):267.
Zaharaki D, Komnitsas K, Perdikatsis V. Use of analytical techniques for identification of inorganic polymer gel composition. J Mater Sci. 2010;45(10):2715–24.
Gasca-Tirado JR, Manzano-Ramírez A, Rivera Muñoz EM, Velázquez-Castillo R, Apátiga-Castro M, Nava R, et al. Ion exchange in geopolymers. In: Selcan K, editor., et al., New trends in ion exchange studies. Rijeka: IntechOpen; 2018.
Virgo D, Mysen BO, Kushiro I. Anionic constitution of 1-atmosphere silicate melts—implications for the structure of igneous melts. Science. 1980;208(4450):1371–3.
Mysen B, Richet P. Silicate glasses and melts. 2nd ed. Amsterdam: Elsevier; 2019.
Steevels IM. Neue erkenntnisse uber die struktur des glases. Philips Tech Rundschau. 1960;9–10:337.
Hench LL. Bioactive ceramics. Ann N Y Acad Sci. 1988;523(1):54–71.
Masson CR. Ionic equilibria in liquid silicates. J Am Ceram Soc. 1968;51(3):134–0.
Hench LL. Glasses and genes: a forecast for the future. Glass Sci Technol. 1997;70:439–48.
Hench LL. Life and death: The ultimate phase transformation. Thermochim Acta. 1996;280:1–13.
Šestak J, Liška M, Hubik P. 12—Oxide glass structure, bridging oxygen and feasible magnetic and structural properties due to the addition of Fe/Mn oxides. In: Sestak J, Mares JJ, Hubik P, editors. Glassy, amorphous and nano-crystalline materials: thermal physics, analysis, structure and properties. Dordrecht: Springer; 2011.
Provis JL, Van Deventer JSJ. Geopolymers: structures, processing, properties and industrial applicatiosn. Sawston: Woodhead Publishing; 2009.
Mazen A, Han-Yong J. Geopolymers and other geosynthetics. Rijeka: Intechopen; 2020.
Davidovits J. Geopolymer chemistry and applications. 5th ed. Saint-Quentin: Geopolymer Institute; 2020.
Kóth J, Sinkó K. Geopolymer composites-in environmentally friendly aspects. Gels. 2023;9:196. https://doi.org/10.3390/gels9030196.
Šestak J, Černy R. Thermotics as an alternative nonequilibrium thermodynamic approach suitable for real thermoanalytical measurements: a short review. J Non-Equilib Thermodyn. 2022;47:233–40.
Plasencia G, Jaramillo D. Thermochemistry in materials processing. Berlin: Springer; 2017.
Delhaes P. Materials and thermodynamics (materials science). Hoboken: Wiley; 2017.
Klimm D. Thermal analysis and thermodynamics in materials science. Berlin: De Gruyter; 2022.
Ebeid EZ, Zakaria M. Thermal analysis: from introductory fundamentals to advanced applications. Amsterdam: Elsevier; 2021.
Acknowledgements
This research has been supported by the Technology Agency of the Czech Republic under project No. FW01010229 and by the specific research project of the University of West Bohemia in Pilsen, NaturTech4 SGS-2022-921.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Šesták, J., Kočí, V., Černý, R. et al. Thirty years since J. Davidovits introduced geopolymers considered now as hypo-crystalline materials within the mers-framework and the effect of oxygen binding: a review. J Therm Anal Calorim 148, 10455–10463 (2023). https://doi.org/10.1007/s10973-023-12312-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-023-12312-z