
Abstract
Two calcined clays (halloysite and kaolinite clays at 700 °C) were used as solid precursors for geopolymer synthesis. This study compares the physicochemical properties of the both resulting geopolymer series heated at 200, 400, 600 and 800 °C. The end specimens were characterized using Optical Dilatometer, XRD, FTIR, MIP and SEM analyses. Results revealed that the flexural strengths were 18.10 and 21.74 MPa for meta-halloysite- and metakaolin-based geopolymers, respectively. After subjected to high temperatures, the flexural strength drastically decreased from 18.10 ± 1.06 to 6.7 ± 0.23 MPa and 21.74 ± 1.20 to 4.63 ± 0.24 MPa, respectively. The maximum shrinkage recorded on metakaolin and meta-halloysite-based geopolymers was 14 and 16% around 950 °C, respectively. The thermal conductivities decreased with increase in heating temperature from 0.78 to 0.19 Wm−1 K−1 and 0.96 to 0.26 Wm−1 K−1, respectively. This reduction is linked to the additional voids and microcracks that occurred within the geopolymer network. The cumulative intrusion in both geopolymers increased with increase in heating temperature up to 600 °C, leading to the degradation of geopolymer network that affected the mechanical strength evolution. Both synthesized geopolymer series are potential candidates for insulation materials or refractory applications.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Alkali-activated binders or geopolymers are cementitious materials formed from a mixture of an aluminosilicate material with media solutions (NaOH, sodium or potassium silicate, phosphoric acid, etc.) at room temperature (< 100 °C) [1,2,3,4,5,6]. Their structure is characterized by tetrahedral coordinated aluminium and silicon atoms bridged with oxygen [7], where the negatively charged aluminium in IV-coordination is compensated by sodium or potassium cations [8]. As reported in the literature, at elevated temperature these materials give better performances and are less destroyed compared with the most used ordinary Portland cement [9,10,11,12,13,14,15]. Numerous studies were carried out and focussed on thermal stability (from room temperature up to 1200 °C) of the geopolymers binders using different aluminosilicates such metakaolin [10, 12, 16], fly ashes [9, 11, 17,18,19] and volcanic ashes [20]. The loss of strength at high temperatures of geopolymers binders depends on diverse parameters like the chemistry and mineralogy of solid precursor and chemical composition of alkaline solution (hardener) used, and solid/liquid ratio during the mixture [21,22,23]. Often the degradation of mechanical strength of geopolymer products exposed at high temperatures is linked to the development of fissures resulting in destruction of amorphous geopolymer binder allowing the better cohesion among different constituents in the matrix. From published works in literature, metakaolin (from calcined kaolin clay in the range of 550–850 °C), composed of reactive alumina and silica as major components, was widely valorized as a key material for the geopolymer synthesis [14, 24, 25]. Previous works [1, 7, 10, 12,13,14, 22, 23, 26] have extensively addressed the fire-resistant of metakaolin-based geopolymer under different conditions synthesis. Duxson et al. [7] explored on the effect of sodium silicate on thermal shrinkage and mass loss of amorphous Na-geopolymer binder derived from metakaolin up to 1200 °C. They found that the thermal shrinkage of resulting samples generally increases with increasing nominal Si/Al ratios. Later, Elimbi et al. [12] studied the thermal behaviour of local Cameroonian metakaolin-based geopolymer heated up to 1000 °C. These authors found that the micrographs of specimens heated from 300 to 900 °C led appear a progressive propagation of microcracks within the matrix and the increased linear shrinkage relating to drastic reduction in compressive strength. Tchakoute et al. [10] prepared sodium alkaline activators using by-products (rice husk ash and waste glass) to produce metakaolin-based geopolymers followed by exposure from 200 to 800 °C. They reported that the total mass loss of geopolymer binders was 13 and 15%, respectively, using sodium water glass from rice husk ash and sodium water glass from waste glass. This was ascribed to the release of structural water at 800 °C. The above-mentioned works and most of those reported in literature were focussed on synthesis conditions and the behaviour of geopolymer binders from metakaolin and by-products (fly ash and slag) heated at high temperatures. The present investigation focussed on the comparative study of the thermal stability up to 800 °C of two prepared geopolymer binders using meta-halloysite from calcined halloysite clay and metakaolin from calcined kaolinite clay used as solid precursors. Halloysite has a structure very similar to that of kaolinite and also belongs to 1:1 dioctahedral clay type except that it contains interlayer water contents between 0 and 2 molecules H2O per Al2Si2O5(OH)4 unit cell [27,28,29,30,31,32,33]. Several previous works focused on using of meta-halloysite obtained from calcined halloysite (550–1000 °C) as a solid precursor for geopolymer synthesis have revealed that the resulting products were amorphous, but depending on the conditions synthesis like hardener used [2, 25, 31, 34,35,36,37], calcination and curing temperatures [25, 30, 38]. Thus, meta-halloysite-based geopolymer products also present high strength and good durability (acidic environment) [30, 39] rendering this clayey material as a potential solid precursor for engineering applications in the same way as metakaolin. The main goal of this research deals with the comparative study of the physicochemical composition and processing conditions on flexural strength, microstructure and heat resistant properties (at temperatures 200–800 °C) of geopolymer binders using meta-halloysite and metakaolin. To make this comparative study effective, the both resulted geopolymer products were characterized using optical dilatometry, X-ray diffractometry (XRD), Fourier transform infrared spectroscopy (FTIR), environmental scanning electron microscope (ESEM) as well as determination of physical properties (flexural strength, thermal shrinkage, water absorption and total pore volume). In addition, the thermal conductivity of both geopolymer series was evaluated in term of heating temperature and interpreted correlating the mechanical strength and microstructure changes.
Experimental
Materials
For this study, two 1:1 dioctahedral phyllosilicate/clay type, i.e. kaolinite and halloysite, were used. They had been collected from Foumban and Balengou, two localities, respectively, situated in the Western Region of Cameroon. Both clay samples were dried in an oven at 105 °C for 48hand ground below 80 µm by dry milling. Then, they were calcined at 700 °C, according to previous work [14]. The resulting powders were labelled MK for calcined kaolin and MH for calcined halloysite.
The commercial sodium silicate solution composed of 14.37 mass% Na2O, 29.54 mass% SiO2 and 56.09 mass% H2O, used in this study was supplied by Ingessil s.r.l., Verona, Italy. The sodium hydroxide pellets (98% of purity, provided by Sigma-Aldrich S.r.l., Milan, Italy), were added to sodium silicate to prepare an alkaline solution with silicate modulus SiO2/Na2O of 1.45 with H2O/Na2O ratio of 10, then stored at room temperature during 24 h before used.
Both meta-halloysite- and metakaolin-based geopolymer binders were prepared by mixing the alkaline solution with different powders in a constant liquid/solid mass of 0.75 in an automatic Hobart mixer for 5 min according to previous work [39]. The different geopolymer pastes were poured into prismatic moulds (140 mm × 10 mm × 10 mm) [40] for 24 h before demoulding. After 28 days curing at room temperature, the samples were divided into five groups with each group containing five samples of each geopolymer series. The first group used as a reference was mechanically tested at 28 days without exposure to high temperatures. The rest of groups (corresponding to second, third, fourth and fifth) were oven dried at 80 °C for 24 h (to avoid an abrupt departure of water out matrix which can cause large voids) and then exposed to 200, 400, 600 and 800°Cat 5 °C.min−1 rate and held at each temperature for 2 h, respectively. The synthesized geopolymer products were labelled GPMH25, GPMH200, GPMH400, GPMH600 and GPMH800 for the specimens made from meta-halloysite and GPMK25, GPMK200, GPMK400, GPMK600 and GPMK800 from metakaolin, respectively. Table 1 presents the detailed summary for the synthesis of the geopolymer binders from both calcined halloysite and kaolinite.
Method
The flexural strengths of geopolymer binders of both geopolymer series were obtained from calcined kaolin and halloysite clays using three-point bending with the Instron® 1195 brand compression machine. The value of flexural strength was calculated from the average of 5 replicated samples for each formulation of both geopolymers series. The standard deviation is presented by errors bar. The flexural strength value of each geopolymer formulation was given by Eq. (1) in accordance with ASTM C1161-13 standard.
where σ is the maximum centre tensile stress (MPa), F maximum load at fracture (N), L is the span, the distance between the supports (mm), b the width, and h the thickness of the specimen (mm).The apparent density and porosity values of geopolymer products were determined according to ASTM C20-00 standard [41]. The water absorption, porosity and apparent density were determined by Archimedes’ method using an electronic balance (Sartorius model 1712001) with a sensitivity of 0 ± 0.001 g. Before carrying the test, the samples were oven cured at 40 °C until constant mass. This principle is based on three measures. First, the sample is weighed in air (Wa) and then in liquid (Wl). Ww the mass of samples after immersion in water. Consequently, the apparent density, water absorption and apparent porosity can be calculated by the following equations.
For the FTIR and XRD analyses, the debris from measurements of the mechanical strength of the geopolymers processed was finely ground and sieved at 60 µm to obtain the powders. For the IR analysis, measurements were made in transmission mode on a Thermo Fisher Scientific Nicolet 380 instrument. To carry out the measurements of each formulation, the powders of each formulation were simply deposited on the diamond before the acquisitions. The data acquisitions were recorded between 4000 and 400 cm−1; the number of scans was 64 with a resolution of 4 cm−1. For data processing and acquisition, the OMNIC software was used. The diffractograms of the raw materials and those of the elaborated geopolymers were obtained on a Brucker-AXS D 5005 apparatus of the Debye-Scherrer type using Cu Kα radiation (λ Kα = 1.54056 Å) and a graphite backside monochromator. The analysis range was between 5 and 70° with a step of 0.04 and an acquisition time of 2 s. The crystalline phases contained in the raw materials and formed products were identified by comparison with the PDF (Powder Diffraction Files) standards from ICDD (International Center for Diffraction Data).
Contactless optical dilatometry (ESS MISURA HSM ODHT model 1600/80, Expert System, Modena, Italy) measurements were performed on both shaped metakaolin and meta-halloysite-based geopolymer binders (5 × 5 × 15 mm3) up to 1300 °C. The measurements were conducted under the same heating conditions (10 °C min−1) for all the samples according to the findings of Kamseu et al. [16].
Collected pieces of both unheated/heated metakaolin and meta-halloysite-based geopolymers obtained from mechanical testing were used for microstructural observation using an Environment Scanning Electron Microscope (ESEM, Quanta200, FEI). The pieces were first mounted in epoxy mould, then polished and coated with carbon prior to SEM observation. Analyses were performed with backscatter electron detector, 15 kV acceleration voltage and about 8 mm working distance.
Pieces collected after the three-point bending tests were used to prepare specimens for the Mercury Intrusion Porosimeter (MIP) (AutoPore IV 9500 V1.09) tests using 1 high-pressure analysis port (228 MPa maximum pressure) and 2 low-pressure analysis ports. Each specimen was put in a penetrometer with 15 mL sample cup and steam volume of 1.1 mL. (The steam volume depends of the penetrometer used for this study used one with steam volume 1.1.) The total pore volume was evaluated using the set-time equilibrium (10 s) mode between pressure limits of 345 kPa and 228 MPa covering the pore diameter range from approximately 0.0055 to 360 µm.
The measurements of thermal conductivity (λ) of both geopolymer GPMHi (i = 25, 200, 600 and 800) and GPMKi (i = 25, 200, 600 and 800) series were carried out using a transient plane source (TPS) technique operating at room temperature and supplied by Hot Disk AB (Sweden). The measurements of each formulation were assessed following the procedure describes by the findings of Bourret et al. [42].
Results and discussion
Characterization of raw materials
The chemical compositions of both solid precursors used in this study are tabulated in Table 2. From Table 2, it is noticed that both kaolin and halloysite clays are mainly composed of (46.20 and 48.0) % SiO2 and (31.30 and 34.0) % Al2O3 as major oxides, respectively. The presence of iron oxide content (8.60%) in raw halloysite justifies the red colour obtained from calcined halloysite clay at 700 °C. Besides, the average particle size of d50 was 8.40 and 9.13 µm, respectively, for kaolin and halloysite clays.
The infrared spectra of both calcined clays (halloysite and kaolinite) are plotted in Fig. 1. The absorption bands appearing at 1045 and 1022 cm−1, respectively, in meta-halloysite (MH) and metakaolin (MK) are attributed to asymmetric Si–O–Si or Si–O–Al bonds stretching, while the ones around 764 and 784 cm−1 correspond to Al–O or Si–O bending. Finally, the last wavenumbers located between 688 and 690 cm−1 are linked to Al–O bending [24, 43].

FTIR spectra of calcined halloysite (MH) and kaolinite (MK) clays
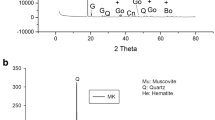
Figure 2a illustrates the X-ray patterns of meta-halloysite (MH) and metakaolin (MK), respectively. Both diffractograms contain similar phases such as quartz (Q) (SiO2, PDF #5-349), haematite (H) (Fe2O3, PDF# 13-5) and anatase (An) (TiO2, PDF #4-447) except for illite (il) (K,H3O)(Al,Mg,Fe)2(Si,Al)4O10[(OH)2,(H2O), PDF # 26-911) that only appears on metakaolin diffractogram. The broad diffraction peaks ranged between 18° and 35°, 2 theta range in both X-ray patterns (Fig. 2a), indicating the formation of amorphous phase after calcination of halloysite and kaolinite clays as required for the alkaline activation. The photographs of unexposed (25 °C) and exposed (200, 400, 600 and 800 °C) geopolymer products from both calcined halloysite and kaolinite are presented in Fig. 2b. It is noticed that without exposure the geopolymers made with meta-halloysite and metakaolin conserved the colour of raw material, dark brown and grey, respectively. The geopolymer samples using meta-halloysite, heated at high temperatures, have seen their colour fading from dark brown to light brown, whereas those from metakaolin, the colour is fading from grey to orange light (Fig. 2b). This change of colour of geopolymer from MK and MH powder is related to the degree of oxidation of irons in the matrix [44, 45]. In addition, the heated specimens GPMH600 and GPMH 800 from meta-halloysite exhibit some microcracks on their surfaces compared to those of GPMK600 and GPMK800 (from metakaolin) that did not show microcracks (Fig. 2b). The appearance of these microcracks is due to the presence of iron minerals that coloured the samples in brown. Thus, the unstable or unreactive iron atoms within the meta-halloysite-based geopolymer matrix exposed above 400 °C, the equilibrium is interrupted in the geopolymer network. This will favour the formation of microcracks, deformation and shrinkage that contributed to slumber the mechanical properties [16].

X-ray patterns of calcined halloysite and kaolinite a and b photographs of unheated and heated geopolymers
Characterization of end products
Phases evolution of geopolymer products
FTIR spectra (Fig. 3a and b) display the molecular bond vibrations of specimens cured at 25 °C and those heated at 200, 600 and 800 °C for the both precursors metakaolin and meta-halloysite, respectively. The shift of larger peaks from 1014 to 968 cm−1 and from 1049 to 979 cm−1 belonging to asymmetric Si–O-Si and/or Al–O–Si stretching for the geopolymer products (GPs) MK and MH, respectively, is evidenced of geopolymerization of Al and Si species from meta-halloysite and metakaolin in alkaline solution [31, 46,47,48,49,50]. The Al(IV) absorption band, typically positioned at around 800 cm−1, superimposed to that attributed to quartz at 798 cm−1 [51], appears in Fig. 3a broad band with maxima at 775 and 779 cm−1 for the MK and MH, respectively, is not existing in the geopolymer samples. In the geopolymers spectra, we can individuate the small sharp peak at about 780 cm−1 attributed to residual quartz in a broader peak shifted to lower wavenumbers around 730 cm−1. In previous studies, the numerous signals of the spectral region 800–550 cm−1ascribed to Secondary Building Units (SBUs), units made of joined SiO4 and AlO4 tetrahedral, forming variously membered rings [48]. These bands are another fingerprint of a geopolymer structure and seem not to be affected by temperature in the case of GPMH series, while the GPMK series shows more evident modification with thermal treatments. In particular, two bands at 779 and 696 cm−1 (all GPMH specimens) could be assigned to asymmetric stretching in Si–O–Al bonds [48]. The less important bands appearing in both infrared spectra in the range of 3345–3386 cm−1 and 1629–1656 cm−1 (Fig. 3a); 3386–3402 cm−1 and 1622–1648 cm−1 (Fig. 3b) are linked to the stretching and bending vibrations of O–H bond belonging to water molecules. These bands diminished with increase in heating from 25 to 800 °C indicating the release of water out of the geopolymer matrix. The last one located between 1388 and 1390 cm−1 (detected on samples GPMK25, GPMK200, GPMH25 and GPMH200) is assigned to vibration modes of carbonate CO32−, providing from the reaction between free sodium and CO2 of atmosphere [19, 52, 53].

FTIR spectra of geopolymer made of calcined halloysite (a) and kaolinite (b)
The XRD patterns of unexposed metakaolin (GPMK25)- and meta-halloysite (GPMH25)-based geopolymers as well as 200, 400, 600 and 800 °C exposed samples (GPMK200, GPMK600 and GPMK800; GPMH200, GPMH600 and GPMH800) are shown in Fig. 4a and b, respectively, for GPMK and GPMH series. These figures exhibited the presence of few crystalline phases such as illite, haematite, quartz and anatase already existing in metakaolin and meta-halloysite X-ray diffractograms. This indicates that these minerals did not likely take part in geopolymerization reaction and also the heating of geopolymer products made from metakaolin and meta-halloysite up to 800 °C did not lead to the new formed crystalline mineral phases. This result quite matches with those of Elimbi et al. [12], Tchakoute et al. [10], Rahier et al. [54] and Rashad et al. [13]. Contrary to the works of Barbosa et al. [55], Sabbatini et al. [56] and Bewa et al. [1] focused on sintered metakaolin-based geopolymers up to 800 °C using different activators. These authors found the formation of nepheline and phosphor-cristobalite in the X-ray pattern of geopolymers. They attributed their presence respectively, to the important amount of Na and Si and polymorphic transformation of mineral berlinite into geopolymer structure. However, the hump peaks belonging to the amorphous seem slightly altered with increasing temperature from room temperature to 800 °C. This likely suggests the reduction or denaturation of geopolymer binder phase, leading to the reduction of the mechanical strengths of the resulting products exposed between 200 and 800 °C [12]. Elimbi et al. [12] showed that when the geopolymer samples were heated at high temperatures, it is noticed the decrease in halo peak that affects the strength development. Elimbi et al. [14] showed that when the geopolymer samples were heated at high temperatures, it is noticed the decrease in halo peak that affect the strength development. Further analysis such as Rietveld refinement analysis needs to be conducted to quantify exactly the amount of amorphous phase in both geopolymer binder series. From 25 to 800 °C, it is observed the reduction in reflection peak of muscovite contained on GPMK samples (Fig. 4a), compared to that of GPMH series. This reduction is attributed to dehydroxylation of muscovite and illite which begins at 650 °C as reported by Mazzucato et al. [57] and Gridi-Bennadji et al. [58]. The beginning dehydroxylation of muscovite could be justified the non-appearance of cracks and fissures or cracks as seen on the micrographs of GPMH series as indicated in Sect. 3.5. Thus, the disorder provided from dehydroxylation of muscovite seems to make the matrix of heated samples GPMK series stronger than GPMH series.

X-ray pattern of geopolymer made of calcined halloysite (a) and kaolinite (b)
Dilatometry and thermal conductivity
Figure 5 shows the sintering behaviour of geopolymers GPMK25, GPMK600, GPMH25 and GPMH600 as collected during heating by the optical dilatometry from 25 to 1200 °C. It can be observed that both samples display very comparable features of thermal shrinkage up to 1000 °C. The GPMK25 and GPMH25 samples exhibited no dimensional change up to 180 °C. From 180 to 380–400 °C, it is noticed a progressive dehydroxylation that allowed the shrinkage of GPMK25 and GPMH25 maintained below 2%, in particular the shrinkage was 2.01 and 2.3%, respectively, GPMK25 and GPMH25. These values remained constant up to 800 and 814 °C, for GPMH25 and GPMK25. The pre-heated samples GPMK600 and GPMH600 specimens exhibited a lower shrinkage around 1%, corresponding to physisorbed water at room temperature before heating. These latter samples remained with stable dimensions up to 812 °C. Similar trend was observed in literature by Duxson et al. [7], Kamseu et al. [16] and Provis et al. [22] between 200 and 600 °C. For these authors, the constant rate of shrinkage in this interval of temperature is related to the physical contraction of geopolymer binder. This is accompanied by the release of water molecules from dehydroxylation of polycondensation of silanol and aluminol oligomers to form network S–O-T(Al) tetrahedral linkage. From Fig. 5, it is not clearly possible to exactly identify the high-temperature expansion peak in each sample, corresponding to the presence of a high-silica gel phase within the geopolymer binder [23]. This is mostly explained by the second derivative curves plotted in Fig. 6a–d. From Fig. 6, the expansion is presented as a small peak between 945 and 1000 °C in all the four studied samples. Such specific high-silica gel expansion phenomenon is concurrent to thermal shrinkage due to the collapse of the structure in terms of densification in presence of liquid phase [16, 22, 55] that accounts for an overall shrinkage of about 14–16%. The highest shrinkages are recorded on geopolymer samples GPMH25 and GPMK25 (cured at room temperature and used as references) compared to GMPH600 and GPMK600. The difference is due by the fact that the latter samples were pre-heated at 600 °C before carried their dilatometry analysis, thus these samples would not much be affected during the dilatometry analysis. The thermal shrinkages recorded on both geopolymer samples, when applying dilatometry analysis, are almost similar to those obtained by Kovárˇík et al. [59] and Tiffo et al. [60] based on pure metakaolin- and kaolin-based geopolymers without reinforcement, respectively. To reduce the shrinkage and microcracks, secondary materials were added fillers sand, alumina powder, etc., leading to the formation of strong matrix [13, 59, 61].

Optical dilatometry curves of geopolymer made of calcined halloysite and kaolinite

Second derivatives curves of geopolymer made of calcined halloysite (a and b) and kaolinite (c and d)
The thermal conductivity (λ) values recorded in both geopolymer series are shown in Table 3. From this table, it is obvious that the thermal conductivity values of unheated samples used as references were 0.78 and 0.96 Wm−1K−1 for GPMH and GPMK specimens. After heating at 800 °C, these values significantly decreased to 0.19 and 0.26 Wm−1K−1, respectively, for GPMH and GPMK geopolymer samples. The high decrease in thermal conductivity is observed when both geopolymer series were heated between 200 and 800 °C. This drastically reduction in thermal conductivity in that interval could be explained by the appearance of voids and crack linked to the destruction of amorphous geopolymer binder responsible for the good cohesion between different components in geopolymer matrix. Hence, this could affect the bulk density geopolymer samples that became lighter due to less dense structure. The comparable tendency was noticed by Sellami et al. [62] who concluded that the decrease in thermal conductivity of geopolymers heated at high temperatures is linked directly proportional to the density of the bulk samples. In similar vein, Kamseu et al. [63, 64], Fongang et al. [65] and Novais et al. [66] also showed that the level of porosity in geopolymer matrix might control the thermal conductivity as well as the mechanical performances. However, the high values observed for GPMK geopolymer series could be related to good cohesion among different particles within the geopolymer matrix that allowed a better heat diffusion through the system.
Flexural strength
The flexural strength behaviour of the MK and MH geopolymers subjected to high temperatures are illustrated in Fig. 7. Without exposure to 200 and 800 °C, the flexural strengths used as references (i.e. at 25 °C, after 28 days curing) were 18.10 ± 1.06 and 21.74 ± 1.20 MPa for GPMH25 and GPMK25 geopolymers, respectively. In term of mechanical performance, a little difference is observed, this could be explained by the impurity within halloysite clay, like haematite, responsible for the red colour exhibited by MH-based geopolymers compared to MK ones which are whiter. This iron mineral (Fe2O3) provides Fe3+ in alkaline solution that can precipitate fast with HO− to form iron hydroxide (Fe(OH)3) [67] either as isolated ions or as oxyhydroxide aggregates. These mixed oxyhydroxides do not react with the starting geopolymer components [20] and can reduce the extension of geopolymer gel within matrix. In addition, such iron-based reactivity could explain the presence of voids or fissures within the matrix, leading to the reduction of mechanical performance. Above 200 °C, it is noted the drastic decrease in flexural strength in both series: for the GPMK and GPMH-based geopolymer series, the strengths were 20.23 ± 1.01, 15.63 ± 0.78, 8.2 ± 0.30 and 6.7 ± 0.23 MPa; 16.70 ± 1.03, 12.30 ± 0.81, 6.20 ± 0.32 and 4.63 ± 0.24 MPa, respectively, at 200, 400, 600 and 800 °C for 2 h. This drastic reduction is due to the loss of water chemically bonded to geopolymer gel structure (N-A-S–H), resulting in the appearance of the microfissures, pores or cracks on the SEM micrographs of samples heated at 600 and 800 °C (see SEM observations paragraph). The trend of mechanical properties obtained in this study quite matches with the observations of Elimbi et al. [12] and Bewa et al. [1] which investigated the thermal behaviour of metakaolin-based geopolymer consolidated, respectively, with alkaline solution and phosphoric acid after subjected to high temperatures, respectively. These authors stated an important strength loss between 400 and 800 °C which is likely linked to departure of hydroxyl groups −OH from the geopolymer network. Although the decline in performances of both geopolymers exposed at high temperatures, the flexural strengths of exposed MK- and MH-based geopolymer binders are still higher when compared to Portland cements reported by Mendes et al. [68] and Martin et al. [69] at the same conditions. Hence, the comparison of the obtained performances in this study with respect to conventional Portland cement shows that the resulting MK and MH-based geopolymers remain valuable materials that are still mechanically resistant after their exposure at elevated temperatures.

Flexural strength of geopolymer of geopolymer made of calcined halloysite and kaolinite
Microstructure
The SEM images describing the internal structure of unheated (GPMK25 and GPMH25 samples) and heated geopolymers (GPMK200, GPMK600 and GPMK800; GPMH200, GPMH600 and GPMH800 specimens) are shown in Figs. 8 and 9, respectively. In Fig. 8a, the micrographs of GPMK25 are compact and very homogeneous justifying the highest gained strength compared to micrographs of GPMH25 which appeared less dense and heterogeneous with the presence of voids and fissures (Fig. 9a). For these samples cured at room temperature, the difference noticed might be clarified by the amount of reactive phase in both solid precursors, during their dissolution in alkaline solution resulting in high polycondensation, leading to high strength. On the other hand, these fissures would also result from water release (occupying the cavity) during the drying of samples with time or by lower bonding strength between different particles in the matrix of GPMH25 sample (Fig. 9a). Therefore, the pronounced formation of voids and fissures within the matrix of GPMH25 geopolymer sample could have contributed to the inhibition of strength development (18.10 ± 1.06 MPa). Even the mechanical properties are not far distanced. After heating, the micrographs of both geopolymer GPMH and GPMK series became most porous, less dense structure with increase in heating temperature from 200 to 800 °C. After exposure to elevated temperatures, i.e. at 600 (Figs. 8b and 9b) and 800 °C (Figs. 8c and 9c), the sintering effect on micrographs of both specimens GPMK600 and GPMH600 showing the appearance of micropores is observed. These pores could be provided by the loss of structural or chemically bound water belonging to geopolymer network. Once more, GPMK600 remains more compact compared to GPMH600. Despite the previously described differences, the decrease in the flexural strength values after heating between 200 and 600 °C was very similar in both geopolymer series, indicating their good cohesion and packing particles effect in the matrix. At 800 °C, GPMH800 appears more deteriorated and porous, whereas GPMK800 sample exhibited minor pores resulting in progressive destruction of amorphous geopolymer binder network leading by poor adhesion between binding phase and other particles (responsible to the high flexural strength obtained from unexposed samples GPMK25 and GPMH25) by increasing the heating temperature resulting in remarkable drop in flexural strength. The trend of mechanical strength (see above) against heating temperature is in agreement with microstructural evolution and well explains the flexural strength development obtained on both geopolymer GPMHi (i = 25, 200, 600 and 800) and GPMKi (i = 25, 200, 600 and 800) series.


SEM images of geopolymer GPMH25 (a), GPMH600 (b) and GPMH800 (c) specimens


SEM images of geopolymer GPMK25 (a), GPMK600 (b) and GPMK800 (c) specimens
Porosity, apparent density and bulk density
The porosity data recorded on both geopolymer series ranged between 32.34–34.40% and 32.83–36.32%, respectively, for GPMHi (i = 25, 200, 600 and 800) and GPMKi (i = 25, 200, 600 and 800) series (Table 2). From Table 2, the porosity increased with increase in heating temperature from 32.01 ± 1.51 to 34.40 ± 2.01% and 32.83 ± 1.72 to 36.32 ± 2.20% up to 600 °C, respectively, for GPMHi (i = 25, 200, 600 and 800) and GPMKi (i = 25, 200, 600 and 800) series. This increase in porosity could be due to voids and pores formation from thermal heating that could retain water molecules once the specimens are immersed. At 800 °C, it decreased from 34.40 ± 2.01 to 33.50 ± 1.43% and 36.32 ± 2.20 to 34.60 ± 1.82%, respectively. The reduction in porosity at 800 °C clearly explains the beginning of sintering effect, leading to limited formation of voids to fix an important content of water within the matrix although their porous nature. The reverse effect was observed on bulk and apparent densities. The bulk density of both geopolymer series increased with rise in heating temperature (25 to 800 °C) from 1.47 ± 0.07 to 1.55 ± 0.08 g cm−3 and 1.43 ± 0.07 to 1.57 ± 0.09 g cm−3, respectively, for GPMHi (i = 25, 200, 600 and 800) and GPMKi (i = 25, 200, 600 and 800) specimens. Similar trend was observed on apparent density determination using MIP analysis (see next paragraph 3.7). The values weakly increased and ranged between 2.16 ± 0.20–2.35 ± 0.12 g cm−3 and 2.20 ± 0.11–2.40 ± 0.15 g cm−3, respectively. The slight increase in both latter parameters could be either due to packing particles effect which positively affected the densification making the samples exposed at higher temperatures a bit heavy.
Mercury intrusion porosimetry
Figure 10a and b depicts the cumulative intrusion against pore size diameter in function of heating temperature. The cumulative intrusion data recorded in both geopolymer series increased with increase in heating temperature from 25 to 600 °C. They ranged between 0.21–0.23 mL g−1 and 0.22–0.25 mL g−1, respectively, for GPMHi (i = 25, 200, 600 and 800) and GPMKi (i = 25, 200, 600 and 800) specimens. After exposure to 800 °C, these values decreased from 0.23 to 0.22 mL g−1 and 0.25 to 0.21 mL g−1, respectively. The increase in cumulative intrusion could be linked to the degradation of geopolymer network caused by the appearance of microcracks and pores that affected the mechanical strength evolution as compared to that of unheated GPMH25 and GPMK25 samples used as references. However, the decrease in cumulative intrusion in both geopolymer series exposed at 800 °C might be due to the beginning of sintering effect accompanied with small densification which seems to limit the propagation of macrovoids within the matrix justifying the lower reduction in flexural strength of both samples heated between 600 and 800 °C as evidenced in Figs. 7, 8c and 9c. This trend is in line with the microstructural evolution against with heating temperature from SEM analysis. The pore sizes distribution of resulting geopolymer products using MIP analysis is reported in Fig. 11a and b for the unheated GPMH25 and GPMK25 geopolymer samples; it ranged between 0.007–0.021 µm and 0.006–0.030 µm, respectively. After heating from 200 to 600 °C, the threshold of pores slightly enlarged from 0.010 to 0.032 µm for GPMH200 and GPMH600 samples, 0.009 to 0.021 µm for GPMK200 and 0.007 to 0.095 µm for GPMK600. This trend confirms the modification occurring in geopolymer structure when they are heated at high temperatures. At 800 °C, the threshold of pores size is more pronounced and ranged between 0.005–0.032 µm and 0.006–0.077 µm, respectively, for GPMH800 and GPMK800 geopolymer specimens. This suggests the important changes that occurred in their structures, leading to the weak obtained mechanical performances compared to other specimens. Meanwhile, it is possible that the samples treated at 800 °C are weak or “soft” with respect to other treated at lower temperatures (25, 200 and 600 °C); in this case, the MIP measure performed at high pressures could provoke a possible pore structure compaction and likely resulted a lower porosity value gained on these samples. Hence, this could limit the propagation of macrovoids or fissures within the matrix although their porous nature. Figure 12a and b exhibits the cumulative pore area data of different geopolymer samples. It is noticed that the cumulative pore area decreased from 60.92 to 28.80 m2 g−1 62.83 to 41.43 m2 g−1 with rise of heating temperature from 25 to 800 °C, respectively, for GPMKi (i = 25, 200, 600 and 800) and GPMHi (i = 25, 200, 600 and 800) series. From these obtained results, it could be concluded that the increase in heating temperature seems to control both the pore size distribution and volume in all studied geopolymer samples.

Cumulative intrusion of metakaolin (a)- and meta-halloysite (b)-based geopolymers

Differential intrusion of metakaolin (a)- and meta-halloysite (b)-based geopolymers

Cumulative pore area of metakaolin (a)- and meta-halloysite (b)-based geopolymers
Permeability and tortuosity prediction based on MIP results theory based on Carniglia paper
Tortuosity factor was calculated considering non-intersecting cylindrical pores, by the following equation [70]:
where Vtot is the total pore volume (cm3 g−1), and ρbulk is the bulk density of sample (g cm−3) conditions that must be fulfilled: 0.05 ≤ Vtot.ρbulk ≤ 0.95.
This correlation is limited to τ values of raging from 1 to 2.2.
The values obtained are raging between 1.82 and 1.87 (Table 2). From Table 3, it is noticed that the tortuosity factor decreased from 1.86 to 1.82 and 1.87 to 1.84 with increase in heating temperature from 25 to 600 °C, respectively, for GMKi (i = 25, 200 and 600) and GMHi (i = 25, 200 and 400) specimens. Afterwards, it increased and reached 1.84 and 1.85 at 800 °C, respectively. It is also noticed that the trend of tortuosity was similar to that of porosity in all geopolymer samples, suggesting that both parameters are linked. Thus, increase in heating temperature from 25 to 800 °C allowed the increase in tortuosity and porosity resulting in limited resistance to diffusion in samples with high porosity. This could limit their use as membrane filtration for waste treatment where high resistance to diffusion is required.
Conclusions
The present project deals with the comparative study of both geopolymer binders made of metakaolin and meta-halloysite in terms of mechanical and microstructural performances. After exposure at high temperatures, the results revealed that both geopolymer series exhibited similar behaviour. From 25 to 800 °C, the progressive microstructural degradation is linked to the reduction in mechanical strength and thermal conductivity of both geopolymer series. The high performances of MK-based geopolymer series compared to that of MH are due to the dehydroxylation of muscovite which contributed to the densification the structure. The slight decrease in cumulative intrusion recorded from MIP data in both geopolymer heated at 800 °C could be either due to packing particles effect or sintering beginning that have positively affected the densification making the samples exposed at this temperature a bit denser. The expansion peaks between 950–1000 °C recorded on dilatometry data quite match strongly with strength development related to destruction of binding phase that allowed this expansion at high temperatures; the total shrinkage ranged 12–14% related to lower extent of crosslinking in binding phase. Both geopolymer products are suitable for thermal applications up to 1000 °C. The obtained excellent properties with both geopolymer series suggest the possibility to design good refractory matrixes through geopolymerization.
References
Nobouassia Bewa C, Tchakouté HK, Fotio D, Rüscher CH, Kamseu E, Leonelli C. Water resistance and thermal behavior of metakaolin-phosphate-based geopolymer cements. J Asian Ceram Soc. 2018;6(3):271–83.
Waijarean N, MacKenzie KJD, Asavapisit S, Piyaphanuwat R, Jameson GNL. Synthesis and properties of geopolymers based on water treatment residue and their immobilization of some heavy metals. J Mater Sci. 2017;52(12):7345–59.
MacKenzie KJD, Brew DRM, Fletcher RA, Vagana R. Formation of aluminosilicate geopolymers from 1:1 layer-lattice minerals pre-treated by various methods: a comparative study. J Materi Sci. 2007;42(12):4667–74.
Tchadjie LN, Ekolu SO. Enhancing the reactivity of aluminosilicate materials toward geopolymer synthesis. J Materi Sci. 2018;53(7):4709–33.
Luukkonen T, Tolonen E-T, Runtti H, Kemppainen K, Perämäki P, Rämö J, Lassi U. Optimization of the metakaolin geopolymer preparation for maximized ammonium adsorption capacity. J Materi Sci. 2017;52(16):9363–76.
Mimboe AG, Abo MT, Djobo JNY, Tome S, Kaze RC, Deutou JGN. Lateritic soil based-compressed earth bricks stabilized with phosphate binder. J Build Eng. 2020;31:101465.
Duxson P, Lukey GC, van Deventer JSJ. Physical evolution of Na-geopolymer derived from metakaolin up to 1000 °C. J Materi Sci. 2007;42(9):3044–54.
Davidovits J. Geopolymers: inorganic polymeric new materials. J Therm Anal Calorim. 1991;37(8):1633–56.
Cheng-Yong H, Yun-Ming L, Abdullah MMAB, Hussin K. Thermal resistance variations of fly ash geopolymers: foaming responses. Sci Rep. 2017;7(1):45355.
Tchakouté HK, Rüscher CH, Kong S, Kamseu E, Leonelli C. Thermal behavior of metakaolin-based geopolymer cements using sodium waterglass from rice husk ash and waste glass as alternative activators. Waste Biomass Valoriz. 2017;8(3):573–84.
Bakharev T. Thermal behaviour of geopolymers prepared using class F fly ash and elevated temperature curing. Cem Concr Res. 2006;36(6):1134–47.
Elimbi A, Tchakoute HK, Kondoh M, Dika Manga J. Thermal behavior and characteristics of fired geopolymers produced from local Cameroonian metakaolin. Ceram Int. 2014;40(3):4515–20.
Rashad AM, Ouda AS. Thermal resistance of alkali-activated metakaolin pastes containing nano-silica particles. J Therm Anal Calorim. 2019;136(2):609–20.
Mierzwiński D, Łach M, Hebda M, Walter J, Szechyńska-Hebda M, Mikuła J. Thermal phenomena of alkali-activated metakaolin studied with a negative temperature coefficient system. J Therm Anal Calorim. 2019;138(6):4167–75.
Adesanya E, Karhu M, Ismailov A, Ohenoja K, Kinnunen P, Illikainen M. Thermal behaviour of ladle slag mortars containing ferrochrome slag aggregates. Advances in Cement Research, 2020, pp. 1–15 (2020).
Kamseu E, Rizzuti A, Leonelli C, Perera D. Enhanced thermal stability in K2O-metakaolin-based geopolymer concretes by Al2O3 and SiO2 fillers addition. J Materi Sci. 2010;45(7):1715–24.
Catauro M, Tranquillo E, Barrino F, Dal Poggetto G, Blanco I, Cicala G, Ognibene G, Recca G. Mechanical and thermal properties of fly ash-filled geopolymers. J Therm Anal Calorim. 2019;138(5):3267–76.
Mustakim SM, Das SK, Mishra J, Aftab A, Alomayri TS, Assaedi HS, Kaze CR. Improvement in fresh, mechanical and microstructural properties of fly ash-blast furnace slag based geopolymer concrete by addition of nano and micro silica. Silicon; 2020.
Assaedi H, Alomayri T, Kaze CR, Jindal BB, Subaer S, Shaikh F, Alraddadi S. Characterization and properties of geopolymer nanocomposites with different contents of nano-CaCO3. Constr Build Mater. 2020;252:119137.
Lemougna PN, MacKenzie KJD, Melo UFC. Synthesis and thermal properties of inorganic polymers (geopolymers) for structural and refractory applications from volcanic ash. Ceram Int. 2011;37(8):3011–8.
Heah CY, Kamarudin H, Mustafa Al Bakri AM, Bnhussain M, Luqman M, Khairul Nizar I, Ruzaidi CM, Liew YM. Study on solids-to-liquid and alkaline activator ratios on kaolin-based geopolymers. Constr Build Mater. 2012;35:912–22.
Provis JL, Harrex RM, Bernal SA, Duxson P, van Deventer JSJ. Dilatometry of geopolymers as a means of selecting desirable fly ash sources. J Non-Cryst Solids. 2012;358(16):1930–7.
Provis JL, Yong CZ, Duxson P, van Deventer JSJ. Correlating mechanical and thermal properties of sodium silicate-fly ash geopolymers. Colloids Surf A. 2009;336(1):57–63.
Nana A, Ngouné J, Kaze RC, Boubakar L, Tchounang SK, Tchakouté HK, Kamseu E, Leonelli C. Room-temperature alkaline activation of feldspathic solid solutions: development of high strength geopolymers. Constr Build Mater. 2019;195:258–68.
Kaze CR, Alomayri T, Hasan A, Tome S, Lecomte-Nana GL, Nemaleu JGD, Tchakoute HK, Kamseu E, Melo UC, Rahier H. Reaction kinetics and rheological behaviour of meta-halloysite based geopolymer cured at room temperature: effect of thermal activation on physicochemical and microstructural properties. Appl Clay Sci. 2020;196:105773.
Boum RBE, Kaze CR, Nemaleu JGD, Djaoyang VB, Rachel NY, Ninla PL, Owono FM, Kamseu E. Thermal behaviour of metakaolin–bauxite blends geopolymer: microstructure and mechanical properties. SN Appl Sci. 2020;2(8):1358.
Lecomte-Nana GL, Lebdioua K, Laffort M, Houta N, Tessier-Doyen N, Abouliatim Y, Peyratout C. Effect of phyllosilicate type on the microstructure and properties of kaolin-based ceramic tapes. In: Developments in Strategic Ceramic Materials II: A Collection of Papers Presented at the 40th International Conference on Advanced Ceramics and Composites, January 24-29, 2016, Daytona Beach, Florida. 2017. Wiley Online Library.
Yuan P, Tan D, Annabi-Bergaya F, Yan W, Fan M, Liu D, He H. Changes in structure, morphology, porosity, and surface activity of mesoporous halloysite nanotubes under heating. Clays Clay Miner. 2012;60(6):561–73.
Yuan P, Tan D, Annabi-Bergaya F. Properties and applications of halloysite nanotubes: recent research advances and future prospects. J Appl Clay Sci. 2015;112:75–93.
Zhang Z, Wang H, Yao X, Zhu Y. Effects of halloysite in kaolin on the formation and properties of geopolymers. J Cem Concr Compos. 2012;34(5):709–15.
Tchakouté HK, Melele SJ, Djamen AT, Kaze CR, Kamseu E, Nanseu CP, Leonelli C, Rüscher CH. Microstructural and mechanical properties of poly (sialate-siloxo) networks obtained using metakaolins from kaolin and halloysite as aluminosilicate sources: a comparative study. Appl Clay Sci. 2020;186:105448.
Joussein E. Geology and mineralogy of nanosized tubular halloysite. In: Developments in Clay Science. 2016, Elsevier. p. 12–48.
Kurczewska J, Pecyna P, Ratajczak M, Gajęcka M, Schroeder G. Halloysite nanotubes as carriers of vancomycin in alginate-based wound dressing. Saudi Pharm J. 2017;25(6):911–20.
Zivica V, Palou MT, Bágeľ TIĽ. High strength metahalloysite based geopolymer. Compos B Eng. 2014;57:155–65.
Bayiha BN, Billong N, Yamb E, Kaze RC, Nzengwa R. Effect of limestone dosages on some properties of geopolymer from thermally activated halloysite. Constr Build Mater. 2019;217:28–35.
MacKenzie K, O’Leary B. Inorganic polymers (geopolymers) containing acid–base indicators as possible colour-change humidity indicators. J Mater Lett. 2009;63(2):230–2.
Zhang B, Guo H, Yuan P, Deng L, Zhong X, Li Y, Wang Q, Liu D. Novel acid-based geopolymer synthesized from nanosized tubular halloysite: the role of precalcination temperature and phosphoric acid concentration. J Cem Concr Compos. 2020;2020:103601.
Zhang B, Guo H, Yuan P, Li Y, Wang Q, Deng L, Liu D. Geopolymerization of halloysite via alkali-activation: dependence of microstructures on precalcination. J Appl Clay Sci. 2020;185:105375.
Kaze CR, Tchakoute HK, Mbakop TT, Mache JR, Kamseu E, Melo UC, Leonelli C, Rahier H. Synthesis and properties of inorganic polymers (geopolymers) derived from Cameroon-meta-halloysite. Ceram Int. 2018;44(15):18499–508.
Kamseu E, Beleukà Moungam LM, Cannio M, Billong N, Chaysuwan D, Melo UC, Leonelli C. Substitution of sodium silicate with rice husk ash-NaOH solution in metakaolin based geopolymer cement concerning reduction in global warming. J Clean Prod. 2017;142:3050–60.
Standard AJWC, PA. ASTM C20-00, Standard test methods for apparent porosity, water absorption, apparent specific gravity, and bulk density of burned refractory brick and shapes by boiling water. 2015.
Bourret J, Tessier-Doyen N, Naït-Ali B, Pennec F, Alzina A, Peyratout CS, Smith DS. Effect of the pore volume fraction on the thermal conductivity and mechanical properties of kaolin-based foams. J Eur Ceram Soc. 2013;33(9):1487–95.
Rees CA, Provis JL, Lukey GC, van Deventer JSJ. The mechanism of geopolymer gel formation investigated through seeded nucleation. Colloids Surf A. 2008;318(1):97–105.
Nazari A, Bagheri A, Dao M, Mallawa C, Zannis P, Zumbo S, Sanjayan JG. The behaviour of iron in geopolymer under thermal shock. Constr Build Mater. 2017;150:248–51.
Bernal SA, Rose V, Provis JL. The fate of iron in blast furnace slag particles during alkali-activation. Mater Chem Phys. 2014;146(1):1–5.
Kaze CR, Venyite P, Nana A, Juvenal DN, Tchakoute HK, Rahier H, Kamseu E, Melo UC, Leonelli C. Meta-halloysite to improve compactness in iron-rich laterite-based alkali activated materials. Mater Chem Phys. 2020;239:122268.
Mozgawa W, Sitarz M. Vibrational spectra of aluminosilicate ring structures. J Mol Struct. 2002;614(1):273–9.
Fernández-Jiménez A, Palomo A. Mid-infrared spectroscopic studies of alkali-activated fly ash structure. Microporous Mesoporous Mater. 2005;86(1):207–14.
Alkan M, Hopa Ç, Yilmaz Z, Güler H. The effect of alkali concentration and solid/liquid ratio on the hydrothermal synthesis of zeolite NaA from natural kaolinite. Microporous Mesoporous Mater. 2005;86(1):176–84.
Wang H, Li H, Wang Y, Yan F. Preparation of macroporous ceramic from metakaolinite-based geopolymer by calcination. Ceram Int. 2015;41(9, Part A):11177–83.
Kljajević LM, Nenadović SS, Nenadović MT, Bundaleski NK, Todorović BŽ, Pavlović VB, Rakočević ZL. Structural and chemical properties of thermally treated geopolymer samples. Ceram Int. 2017;43(9):6700–8.
Kaze CR, Djobo JNY, Nana A, Tchakoute HK, Kamseu E, Melo UC, Leonelli C, Rahier H. Effect of silicate modulus on the setting, mechanical strength and microstructure of iron-rich aluminosilicate (laterite) based-geopolymer cured at room temperature. Ceram Int. 2018;44(17):21442–50.
Sontia Metekong JV, Kaze CR, Deutou JG, Venyite P, Nana A, Kamseu E, Melo UC, Tatietse TT. Evaluation of performances of volcanic-ash-laterite based blended geopolymer concretes: mechanical properties and durability. J Build Eng. 2020;2020:101935.
Rahier H, Wastiels J, Biesemans M, Willlem R, Van Assche G, Van Mele B. Reaction mechanism, kinetics and high temperature transformations of geopolymers. J Materi Sci. 2007;42(9):2982–96.
Barbosa VF, MacKenzie KJ. Thermal behaviour of inorganic geopolymers and composites derived from sodium polysialate. J Mater Res Bull. 2003;38(2):319–31.
Sabbatini A, Vidal L, Pettinari C, Sobrados I, Rossignol S. Control of shaping and thermal resistance of metakaolin-based geopolymers. Mater Des. 2017;116:374–85.
Mazzucato E, Artioli G, Gualtieri AJP, C.o. Minerals, High temperature dehydroxylation of muscovite-2M1: a kinetic study by in situ XRPD. 1999;26:375–81.
Gridi-Bennadji F, Beneu B, Laval JP, Blanchart P. Structural transformations of Muscovite at high temperature by X-ray and neutron diffraction. Appl Clay Sci. 2008;38(3):259–67.
Kovářík T, Rieger D, Kadlec J, Křenek T, Kullová L, Pola M, Bělský P, Franče P, Říha J. Thermomechanical properties of particle-reinforced geopolymer composite with various aggregate gradation of fine ceramic filler. Constr Build Mater. 2017;143:599–606.
Tiffo E, Bike Mbah JB, Belibi Belibi PD, Yankwa Djobo JN, Elimbi A. Physical and mechanical properties of unheated and heated kaolin based-geopolymers with partial replacement of aluminium hydroxide. Mater Chem Phys. 2020;239:122103.
Van Riessen A, Rickard W, Sanjayan J. 15-Thermal properties of geopolymers. In: Provis JL, van Deventer JSJ, editors. Geopolymers. New York: Woodhead Publishing; 2009. p. 315–42.
Sellami M, Barre M, Toumi M. Synthesis, thermal properties and electrical conductivity of phosphoric acid-based geopolymer with metakaolin. Appl Clay Sci. 2019;180:105192.
Kamseu E, Ngouloure ZNM, Ali BN, Zekeng S, Melo UC, Rossignol S, Leonelli C. Cumulative pore volume, pore size distribution and phases percolation in porous inorganic polymer composites: relation microstructure and effective thermal conductivity. Energy Build. 2015;88:45–56.
Kamseu E, Nait-Ali B, Bignozzi MC, Leonelli C, Rossignol S, Smith DS. Bulk composition and microstructure dependence of effective thermal conductivity of porous inorganic polymer cements. J Eur Ceram Soc. 2012;32(8):1593–603.
Fongang RTT, Pemndje J, Lemougna PN, Melo UC, Nanseu CP, Nait-Ali B, Kamseu E, Leonelli C. Cleaner production of the lightweight insulating composites: microstructure, pore network and thermal conductivity. Energy Build. 2015;107:113–22.
Novais RM, Buruberri L, Ascensão G, Seabra M, Labrincha J. Porous biomass fly ash-based geopolymers with tailored thermal conductivity. J Clean Prod. 2016;119:99–107.
van Deventer JSJ, Provis JL, Duxson P, Lukey GC. Reaction mechanisms in the geopolymeric conversion of inorganic waste to useful products. J Hazard Mater. 2007;139(3):506–13.
Mendes A, Sanjayan J, Collins F. Phase transformations and mechanical strength of OPC/Slag pastes submitted to high temperatures. Mater Struct. 2007;41(2):345.
Martin A, Pastor JY, Palomo A, FernándezJiménez A. Mechanical behaviour at high temperature of alkali-activated aluminosilicates (geopolymers). Constr Build Mater. 2015;93:1188–96.
Carniglia SC. Construction of the tortuosity factor from porosimetry. J Catal. 1986;102(2):401–18.
Acknowledgements
RCK grateful acknowledges the financial support of AVRUL (Agence pour la Valorisation de la Recherche Universitaire du Limousin) for his stay in IRCER, UMRCNRS 7315, Limoges under 2018 AVRUL postgraduate internship. This project received the contribution of the FLAIR fellowship African Academic of Science and the Royal Society through the funding N° FLR/R1/201402. The authors also are grateful to Ingessil S.r.l., Verona, Italy, for providing sodium silicate used.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kaze, C.R., Nana, A., Lecomte-Nana, G.L. et al. Thermal behaviour and microstructural evolution of metakaolin and meta-halloysite-based geopolymer binders: a comparative study. J Therm Anal Calorim 147, 2055–2071 (2022). https://doi.org/10.1007/s10973-021-10555-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-021-10555-2