
Abstract
In this study, a complex oxide solid solution consisting of lanthanum gallate with partial substitutions for strontium and magnesium (La0.9Sr0.1Ga0.8Mg0.2O2.85) was synthesized by the sol–gel route, aiming to obtain a sinter active powder and a final material that could be stoichiometrically controlled. The thermal behavior of the synthesized powder involves several steps of decomposition. The linear shrinkage of green compacts up to 1500 °C was 31%. The bulk conductivity of sintered specimens increases with sintering temperature up to 1400 °C. The stoichiometry was maintained for sintering temperatures up to 1450 °C. Elemental mapping obtained by energy-dispersive spectroscopy evidenced magnesium segregation at the grain boundaries. The overall results evidence the suitability of the synthesis method for preparing doped lanthanum gallate.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Mixed oxides and complex oxide solid solutions consisting of several cations are receiving great attention due to their unique and special properties. Examples of these materials may be found in diverse areas such as biotechnological and biomedical applications [1], catalysis [2], renewable energy [3, 4], and battery development [5].
Lanthanum gallate with partial substitutions for strontium and magnesium, La1−xSrxGa1−yMgyO3−δ, where 0.1 ≤ x, y ≤ 0.2, and perovskite structure is a promising material as solid electrolyte and electrode in solid oxide fuel cells operating at intermediate temperatures [4, 6, 7]. High ionic conductivity, low electronic conductivity, and wide electrolytic domain are the main properties of this family of oxide-ion conductors [4, 6, 7].
The mixing of starting reagents followed by high-temperature reaction is often employed for preparing doped lanthanum gallate [6,7,8,9,10]. This method offers some advantages, including high production rate and moderate cost, although undesired phases, mainly La4Ga2O9, LaSrGaO4, and LaSrGa3O7, are frequently observed [9, 11, 12]. The physicochemical properties of these materials depend on the method of synthesis [13]. The electrical resistance measured in solid oxide fuel cells consisting of lanthanum gallate is largely influenced by changes in the compound stoichiometry and manufacturing process [14]. In this context, solution methods are preferred for the synthesis of doped lanthanum gallate because they enable minimization of undesired phases and better stoichiometry control.
Previous studies applying solution methods for preparing doped lanthanum gallates focused on variants of simultaneous precipitation (also called coprecipitation), such as coprecipitation starting with acetate precursors [15], carbonate coprecipitation [16], freeze drying following coprecipitation [17], and combustion [13, 18]. Few works may be found utilizing solution techniques where a polymer, an amorphous, or a gelatinous matrix is employed [19,20,21]. These latter methods are preferred for synthesizing complex compounds due to the relatively low diffusion rate of cations in those matrices, allowing for their better distribution without or with negligible segregation [22].
In this work, the compound La0.9Sr0.1Ga0.8Mg0.2O2.85, hereafter LSGM, was synthesized by a soft chemistry route, with the aim of minimizing the fraction of undesired phases and obtaining high densification with homogeneous microstructure and stoichiometry control.
Experimental
Materials and sample preparation
The La0.9Sr0.1Ga0.8Mg0.2O2.85 nominal composition was synthesized using a sol–gel method based on that proposed in [23]. Similar methods have been used for studying the sintering behavior of LSGM either by the conventional or fast firing [20, 24], and other simple [25] or complex oxides [26]. The main differences between these methods are briefly described below.
Lanthanum nitrate (99.99%, Alfa Aesar), gallium nitrate (99.99%, Alfa Aesar), and strontium carbonate (99%, Sigma Aldrich) and magnesium nitrate (99%, Sigma Aldrich) were used as starting materials without further purification. Stock solutions of the starting nitrates were prepared with distilled water. The strontium nitrate solution was prepared by dissolving the starting strontium carbonate in nitric acid 36% (P.A., Sigma-Aldric) by dipping nitric acid onto the carbonate solution. The concentration of the stock solutions was determined by gravimetry and fixed at 1 mol L−1. Stoichiometric amounts of the stock solutions were mixed under magnetic stirring and heating for homogenization. Afterward, an aqueous solution of anhydrous citric acid, C6H8O7 (P. A.-ACS, Synth), was added to the cation solution. In general, 1 mol of citric acid is sufficient for complexation of cations in solution. In this case, the citric acid/metal ratio was set to 3:1 to avoid precipitation of the strontium precursor. The temperature of the resulting solution was raised to 70 °C, promoting gradual elimination of water and NOx vapor. Continuous heating of the clean and transparent cation solution produced a yellow porous foam. The foam was heat-treated at 250 °C for 1 h and deagglomerated in an agate mortar with pestle. Calcination of the heat-treated foam was accomplished at 800 °C for 1 h. All thermal treatments were completed in air with furnace cooling, contrary to what was previously proposed [23].
Disc-shaped samples (ϕ 8 mm and 2 mm thickness) were obtained by uniaxially pressed (20 MPa) followed by cold isostatic pressing at 100 MPa. Sintering experiments were conducted between 1300 and 1500 °C for 4 h, and at 1400 °C for 4, 10, and 15 h.
Characterization
About 50 mg of the porous foam without any previous heat treatment was characterized by simultaneous thermogravimetry (TG) and differential thermal analysis (DTA; Netzsch, STA409E) up to 1000 °C, heating at a rate of 10 °C min−1, under synthetic air (5 mL min−1), with α-alumina as the reference material. Structural characterization of powders and sintered samples was performed by X-ray diffraction (XRD) (Bruker-AXS, D8 Advance) at 40 kV and 40 mA, Ni-filtered Cu Kα radiation (λ = 1.5405 Å) in the 20° ≤ 2θ ≤ 80° range, with 0.05° step size. The diffraction patterns were indexed with the corresponding diffraction files. The linear shrinkage of calcined and pressed (green compacts) specimens was measured by thermodilatometry (Anter, Unitherm 1161™) up to 1500 °C with a 10 °C min−1 heating rate. True elemental composition of sintered samples was determined by X-ray fluorescence (XRF, Shimadzu, EDX-720). The crystallite size was estimated by the Debye–Scherrer equation. The apparent density was measured using the water immersion method allowing for calculating the relative density of sintered samples. The theoretical density used was 6.67 g cm−3 (ICSD 51-288).
Polished and thermally etched surfaces of samples were observed by scanning electron microscopy (SEM, Philips, XL30) for topography analysis, coupled to energy-dispersive X-ray analysis (EDS, EDAX, Apollo 10), for elemental mapping. The mean grain size was determined by the intercept method. Additional spectroscopic characterization was performed by measurement of the ionic conductivity by electrochemical impedance spectroscopy (HP 4192A). Silver was used as the electrode for measurements in the 5 to 13 MHz and 280 to 420 °C frequency and temperature ranges, respectively, with 100 mV of applied (AC) voltage.
Results and discussion
The thermal decomposition behavior of the porous foam before any heat treatment is depicted in Fig. 1. The TG curve shows total mass loss of about 81% up to 650 °C. The mass loss occurs in several steps with corresponding thermal events in the DTA curve. The first step occurs up to approximately 350 °C, with mass loss of 64% and endothermic peaks at 145 °C and about 240 °C as main thermal events. The first endothermic peak is associated with foam dehydration, whereas the second peak is related to the thermal decomposition of free citric acid [27], added in excess during synthesis.

a Thermogravimetry TG and b DTA curves of the porous foam
The second step of mass loss (14%) extending from 350 to about 500 °C is accompanied by a large exothermic peak with maximum at 355 °C with shoulders at 400 and 450 °C. These several peaks occurring during the thermal decomposition of the porous foam are probably related to the complex decomposition of the mixed citrate. The lanthanum citrate, for example, is known to decompose involving intermediates, such as lanthanum carbonate and oxycarbonate [28]. This step is attributed to the simultaneous decomposition of organic matter, CO and CO2, and burnout of the decomposition products. The final step of mass loss of about 3%, from about 500 to 650 °C, is assigned to the final combustion of the residual decomposition products and crystallization of LSGM, with an exothermic peak at 635 °C.
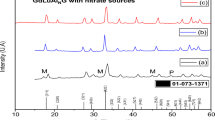
To determine the nature of the DTA peaks, some XRD experiments were performed after heating powder samples up to specific temperatures followed by cooling to room temperature without a holding time (Fig. 2). The as-prepared and the heat-treated (250 °C for 1 h) foams are amorphous to X-rays. Calcination of the as-prepared foam at 800 °C led to the desired composition (indicated by *), along with undesired phases (indicated by numbers 1, 2 and 3, La4Ga2O9, LaSrGa3O7 and SrO, respectively), due to the incomplete reaction among the oxides at such a low temperature. That temperature is about 400 °C lower than that usually employed in the synthesis of doped lanthanum gallate via the conventional solid-state reaction method [9], and 200 °C lower than nanopowders synthesized via other solution methods [13, 16, 19]. These results emphasize the decisive role of this synthesis method for preparing fine particulate materials with high homogeneity.

XRD patterns of the porous foam after specific thermal treatments. 1-La4Ga2O9, 2-LaSrGa3O7, 3-SrO and (*) LSGM
Figure 3 shows a SEM micrograph of the calcined powder. The main microstructure features are agglomerated particles with wide size distribution. The crystallite size estimated by Debye–Scherrer is about 40 nm.

SEM micrograph of the calcined powder
The linear shrinkage of green compacts up to 1500 °C and the first derivative curves are depicted in Fig. 4. The shrinkage starts at approximately 900 °C, accounting for a 31% shrinkage at the final experimental temperature. Two shrinkage stages were observed at temperatures corresponding to the maximum shrinkage rates: 1100 °C in the first stage (from ~ 900 to ~ 1140 °C) and 1420 °C in the second stage (from ~ 1140 to 1500 °C). The detection of the two stages in the linear shrinkage curve of LSGM may be attributed to several factors, such as agglomerates, as observed in the SEM micrograph (Fig. 3), phase changes, impurities and changes in the main sintering mechanism, among others. It is worth to note that for the same compound prepared by solid-state route, the final shrinkage achieved was only 18% under the same experimental conditions [9].

Linear shrinkage and first derivative curves of green compacts
Table 1 lists relative density values obtained for specimens sintered at different temperatures and for several holding times. A steady increase in the density was observed with both temperature and holding time. For all sintering temperature and time profiles, the LSGM specimens achieved relatively high densities (higher than 92%).
The XRD patterns of the samples sintered at several temperatures for four hours (Fig. 5a) and for different holding times at 1400 °C (Fig. 5b) show narrow diffraction peaks, all of them belonging to a single-phase LSGM with the characteristic orthorhombic structure. There is no indication of undesired phases, even for relatively low sintering temperatures (1300 °C). The diffraction peaks were indexed according to the ICSD 51-288 file of the orthorhombic structure of LSGM. The gradual increase in the bulk conductivity of LSGM seems to be related to the density of the sintered specimen, because at 1300 °C the orthorhombic phase (space group Imma and cell parameters 5.5179(1) 7.8200(2) 5.5394(1) Å—ICSD 51-288) had already been formed (Fig. 5).

XRD patterns of LSGM sintered at several a temperatures and b times
The [–Z′′ (ω) × Z′ (ω)] impedance spectroscopy diagrams of LSGM specimens sintered at several temperatures are shown in Fig. 6. The numbers over the experimental points in these plots are the decimal logarithm of the frequency. The real [Z′ (ω)] and the imaginary [–Z′′ (ω)] components of the total impedance were normalized for the geometry of the specimens for the purpose of comparison.

Electrochemical impedance spectroscopy diagrams of LSGM specimens sintered at several temperatures. Inset: Expanded view of the high-frequency range
For polycrystalline ceramics with ion-conducting behavior, the impedance diagrams consist of a sequence of decentralized arcs identified with the resistive and capacitive effects of the bulk (or grains) at high frequencies, grain boundaries (intermediate frequencies), and the reactions occurring at the electrode/electrolyte interface (low frequencies) [29]. The high-frequency range in Fig. 6 is highlighted in the inset, revealing differences in the bulk properties with different sintering temperatures. The blocking of charge carriers (oxide-ion vacancies), usually detected at intermediate frequencies, is not seen in these plots, probably due to the relatively large grain sizes (Table 1) of LSGM specimens as a consequence of the high sintering temperatures. In general, the larger the grain size, the smaller the grain boundary blocking effect in ionic conductors.
The temperature dependence of the intragrain conductivity of sintered LSGM is shown in Fig. 7. The effect of the holding time was investigated for specimens sintered at 1400 ºC exhibiting high ionic conductivity (Fig. 7a). As can be seen (Fig. 7a) no substantial differences were observed in the bulk conductivity. This means that all the thermal energy supplied to the ceramic at the sintering temperature was used for densification and grain growth, without any change in the stoichiometry. The bulk conductivity of specimens sintered at different temperatures (Fig. 7b) increased up to approximately 1400 °C, and decreased for the highest sintering temperature (1500 °C). The gradual increase in the bulk conductivity of LSGM seems to be related to the density of the sintered specimen, because at 1300 °C the orthorhombic phase had already formed (Fig. 5). In this context, the minimum density of the sintered specimens should be higher than 92% of the theoretical value to achieve high conductivity. That decrease in the ionic conductivity for sintering at 1500 °C may be related to deviation from stoichiometry. The apparent activation energy for conduction is 1.01 ± 0.05 eV. The specimen sintered at 1500 °C shows a slightly higher (1.08 ± 0.05 eV) activation energy.

Temperature dependence of the ionic conductivity of LSGM specimens sintered at different a holding times and b temperatures
The following experiments were conducted on specimens sintered at 1450 °C for four hours. The elemental composition determined by XRF measurements is summarized in Table 2 along with nominal values. The determined elemental fractions agree, within experimental errors, with the nominal values, ensuring the desired stoichiometry. This is an additional advantage of this method whenever complex compounds are synthesized. Loss of gallium in the form of Ga2O is thought to occur during thermal treatments at high temperatures [30]. Results in Table 2 suggest no Ga loss for sintering temperatures up to 1450 °C.
Figure 8 shows SEM micrographs obtained for specimens sintered at 1350 and 1400 °C for 4 h. The main microstructure features are micrometric grains and homogeneity with respect to grain size and morphology. The increase in temperature promoted the grain growth and the gradual decrease of open porosity. The mean grain size of sintered specimens estimated by the intercept method is listed in Table 1. Increasing both dwell temperature and time promoted grain growth, as expected, reaching values higher than 10 μm for sintering at 1450 °C for 4 h.

SEM micrograph of the LSGM specimen sintered for 4 h at: a 1350 ºC and b 1400 °C
Figure 9 shows the microstructure characterization of LSGM by SEM coupled to EDS analysis. The micrograph detected with backscattered electrons (Fig. 9a) revealed some grain pullout and porosity concentrated at grain boundaries. The elemental mapping in Fig. 9b evidences a random and homogeneous distribution of La (purple), Sr (green), and Ga (blue) in the matrix. No indication of segregation of those elements was observed, contrary to what was reported for specimens prepared by solid-state synthesis [9]. This result agrees with the XRD result (Fig. 5a). Figure 9c highlights the elemental map of Mg (red). The elemental distribution is random over the observed microregion, although non-homogenous and with a high concentration of Mg at the grain boundaries. Mg precipitation at the grain boundaries was not detected by XRD due to the experimental limitations of the technique, but it was already evidenced by dark contrast grains appearing in SEM micrographs [31, 32], and seems to be related to the thermodynamics of phase formation in this complex solid solution.

SEM micrograph and EDS analysis of the LSGM specimen sintered at 1450 ºC for 4 h: a backscattered image, b, c elemental mapping of La (purple), Sr (green), Ga (blue), and Mg (red). (Color figure online)
These overall results show that stoichiometric complex oxide solid solutions may be easily synthesized using this solution method, allowing for the production of high-density ceramic compacts with minimization of undesired phases.
Conclusions
A solution method using an amorphous matrix was successfully applied to synthesize doped lanthanum gallate. The thermal decomposition of the synthesized porous foam was almost complete at 650 °C. Crystallization of the LSGM phase shows an exothermic peak at 635 °C. The linear shrinkage of green compacts was 31% up to 1500 ºC. Relative density values of sintered specimens were in 92–96% for sintering temperatures of 1300–1500 °C. High ionic conductivity was obtained for specimens sintered at 1400 °C. XRF results evidenced the suitability of the powder preparation method to control the stoichiometry of the produced bulk specimens.
References
Singh DP, Herrera CE, Singh B, Singh S, Singh RK, Kumar R. Graphene oxide: an efficient material and recent approach for biotechnological and biomedical applications. Mater Sci Eng C Mater Biol Appl. 2018;86:173–97.
Grant JT, Venegas JM, McDermott WP, Hermans I. Aerobic oxidations of light alkanes over solid metal oxide catalysts. Chem Rev. 2018;118:2769–815.
Sivula K, Van de Krol R. Semiconducting materials for photoelectrochemical energy conversion. Nat Rev Mater. 2016;1:15010.
Sunarra J, Hashim SS, Zhu N, Zhou W. Perovskite oxides applications in high temperature oxygen separation, solid oxide fuel cells and membrane reactor: A review. Progr Energy Combust Sci. 2017;61:57–77.
Zhao Y, Li XF, Yan B, Xiong DB, Li DJ, Lawes S, Sun L. Recent developments and understanding of novel mixed transition-metal oxides as anodes in lithium ion batteries. Adv Energy Mater. 2016;6:1502175.
Ishihara T, Honda M, Takita Y. Doped LaGaO3 perovskite type oxide as a new oxide ionic conductor. J Am Chem Soc. 1994;116:3801–3.
Feng M, Goodenough JB. A superior oxide-ion electrolyte. Eur J Solid State Inorg Chem. 1994;31:663–72.
Li M, Zhang Y, An M, Lü Z, Huang X, Xiao J, Wei B, Zhu X, Su W. Synthesis and characterization of La0.9Sr0.1Ga0.8Mg0.2O3−δ intermediate temperature electrolyte using conventional solid state reaction. J Power Sourc. 2012;218:233–6.
Reis SL, Muccillo ENS. Preparation of dense La0.9Sr0.1Ga0.8Mg0.2O3−δ with high ionic conductivity by solid-state synthesis. Ionics. 2018;24:1693–700.
Zhang X, Liu T, Zhang H. Limiting current oxygen sensors with La0.8Sr0.2Ga0.8Mg0.2O3−δ electrolyte and La0.8Sr0.2(Ga0.8Mg0.2)1−xCoxO3−δ dense diffusion barrier. Ionics. 2018;24:827–32.
Rozumek M, Majewski P, Aldinger F, Künstler K, Tomandl G. Preparation and electrical conductivity of common impurity phases in (La, Sr)(Ga, Mg)O3 solid electrolytes. CFI-Ceramic Forum Int/Ber D Keram Ges. 2003;80:E35–E40.
Djurado E, Labeau M. Second phases in doped lanthanum gallate perovskites. J Eur Ceram Soc. 1998;18:1397–404.
Chaubey N, Wani BN, Bharadwaj SR, Chattopadhyaya MC. Influence of synthesis route on physicochemical properties of nanostructured electrolyte material La0.9Sr0.1Ga0.8Mg0.2O3−δ for IT-SOFCs. J Therm Anal Calorim. 2013;112:155–64.
Wendel CH, Gao Z, Barnett SA, Braun RJ. Modeling and experimental performance of an intermediate temperature reversible solid oxide cell for high-efficiency, distributed-scale electrical energy storage. J Power Sourc. 2015;283:329–42.
Huang K, Feng M, Goodenough JB. Wet chemical synthesis of Sr- and Mg-doped LaGaO3, a perovskite-type oxide-ion conductor. J Am Chem Soc. 1996;79:1100–4.
Cristiani C, Zampori L, Latorrata S, Pelosato R, Dotelli G, Ruffo R. Carbonate coprecipitation synthesis of Sr- and Mg-doped LaGaO3. Mater Lett. 2009;63:1892–4.
Marrero-López D, Martin-Sedeño MC, Peña-Martínez J, Ruiz-Morales JC, Ramos-Barrado JR. Microstructure and conductivity of La1−xSrxGa0.8Mg0.2O3−δ electrolytes prepared using the freeze-drying method. J Am Ceram Soc. 2011;94:1031–9.
Yu S, Bi H, Sun J, Zhu L, Yu H, Lu C, Liu X. Effect of grain size on the electrical properties of strontium and magnesium doped lanthanum gallate electrolytes. J Alloy Compd. 2019;777:244–51.
Singh RK, Singh PS. Synthesis of La0.9Sr0.1Ga0.8Mg0.2O3−δ electrolyte via ethylene glycol route and its characterizations for IT-SOFC. Ceram Int. 2014;40:7177–84.
Polini R, Pamio A, Traversa E. Effect of synthetic route on sintering behavior, phase purity and conductivity of Sr- and Mg-doped LaGaO3 perovskites. J Eur Ceram Soc. 2004;24:1365–70.
Shi M, Xu Y, Liu A, Liu N, Wang C, Majewski P, Aldinger F. Synthesis and characterization of Sr- and Mg-doped lanthanum gallate electrolyte materials prepared via the Pechini method. Mater Chem Phys. 2009;114:43–6.
Johnson DW Jr. Non-conventional powder preparation techniques. Am Ceram Soc Bull. 1981;60:221–2.
Marcilly C, Courty P, Delmon B. Preparation of highly dispersed mixed oxides and solid solutions by pyrolysis of amorphous precursors. J Am Ceram Soc. 1970;53:56–7.
Reis SL, Muccillo ENS. Ionic conductivity of chemically synthesized La0.9Sr0.1Ga0.8Mg0.2O3−δ solid electrolyte. Adv Mater Res. 2014;975:81–5.
Romanova I, Kirillov S. Preparation of Cu, Ni and Cooxides by a citric acid-aided route. J Therm Anal Calorim. 2018;132:503–12.
Porfirio TC, Muccillo ENS. Thermal and electrical properties of CaCu3Ti4O12 synthesized by soft chemistry route. J Therm Anal Calorim. 2018;133:851–7.
Wiecinska P. Thermal degradation of organic additives used in colloidal shaping of ceramics investigated by the coupled DTA/TG/MS analysis. J Therm Anal Calorim. 2016;123:1419–30.
Wu S, Chang Z, Wang K, Xiong W. Preparation and thermal behaviour of rare earth citrate hydrates. J Therm Anal. 1995;45:199–206.
Bauerle JE. Study of solid electrolyte by a complex admittance method. J Phys Chem Solids. 1969;30:2657–70.
Rozumek M, Majewski P, Sauter L, Aldinger F. Homogeneity region of strontium-and magnesium-containing LaGaO3 at temperatures between 1100ºC and 1500ºC in air. J Am Ceram Soc. 2003;86:1940–6.
Oncel C, Ozkaya B, Gulgun MA. X-ray single phase LSGM at 1350 ºC. J Eur Ceram Soc. 2007;27:599–604.
Zhao X, Li X, Xu N, Huang K. Beneficial effects of Mg-excess in La1−xSrxGa1−yMgy+zO3−δ as solid electrolyte. Solid State Ionics. 2012;214:56–61.
Acknowledgements
The authors acknowledge FAPESP (2013/07296-2), CNPq (305889/2018-4), CAPES (Finance Code 001) and CNEN for financial supports, and the Laboratory of Electron Microscopy and Microanalysis (LMM/CECTM) at IPEN for SEM/EDS analyses.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Reis, S.L., Grosso, R.L. & Muccillo, E.N.S. Thermal and spectroscopic characterization of sol–gel-synthesized doped lanthanum gallate. J Therm Anal Calorim 146, 1561–1567 (2021). https://doi.org/10.1007/s10973-020-10113-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-020-10113-2