
Abstract
The crystallization evolution of lead zirconate titanate (PZT) powders processed from two organic gel systems with nominal composition was investigated. The crystallization of the perovskite PZT form initiated as the calcining temperature reached 400 °C. The formation of PbO phase constituted seeding grains for PZT crystallization at such low temperature. The formation of lead-deficient fluorite or pyrochlore phases was completely avoided under judicious processing conditions. TGA/DTA analysis was carried out to evaluate the activation energy associated with the perovskite phase formation. The kinetic parameters of the PZT crystallization toward perovskite formation were followed by employing Kissinger and JMAK isoconversional kinetic models. The results revealed that the crystallization mechanism is related to the powder precursor system. The value of the activation energy for the perovskite formation was about 77 kJ mol−1 irrespective of the precursor system when the Kissinger model was applied.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Lead zirconate titanates are of great technological importance due to their excellent ferroelectric and piezoelectric properties. PZT-based materials are commonly applied in electronics and microelectronics. The best piezoelectric properties are associated with the morphotropic composition.
The synthesis of PZT powder of high quality is essential to develop superior piezoelectric PZT ceramics. PZT powders are mostly prepared through conventional solid-state reaction starting from their individual metal oxides. Materials synthesized via this technique are often very fragile and porous to be easily polarized [1]. Chemical processing routes are usually used to maintain the stoichiometry and to lower the crystallization temperature and particle size in order to produce highly dense product.
The fabrication of high-quality PZT powders at low temperature is highly required. The crystallization of the PZT perovskite phase is compelled by nucleation, which needs considerable activation energy [2]. The kinetic analysis of the thermal decomposition route is an essential step to comprehend how the material behaves under different functioning conditions. In addition, the determination of the kinetic mechanism helps optimizing the processing parameters. The kinetics of sol–gel reactions has been explored by several groups. Fernandes et al. [3] have found that the rate of PZT crystallization is directly related to the decline of the amorphous phase content, which is in turn affected by the oxidation of the carbonaceous mass. Merckle et al. [4] investigated isothermal and non-isothermal crystallization of PZT (45/55) sample processed by the sol–gel method using 2-methoxyethanol as the solvent. They reported an apparent activation energy around 300 kJ mol−1 for the pyrochlore-to-perovskite conversion and an Avrami exponent of 1.
This study aims to investigate the structural evolution and the crystallization kinetics in order to determine the most plausible growth mechanism of the sol–gel-processed PZT nanopowders. Two sol precursor systems will be explored. The main objective of this work is to investigate the crystallization behavior of PZT powder and to determine important kinetic parameters related to the perovskite phase formation. The DTA peak corresponding to the perovskite crystallization was analyzed by two theoretical models, which have been formulated by Kissinger and JMAK, to clarify the nucleation and growth processes.
Experimental
The procedures used to prepare these precursor systems were previously described in detail [5, 6]. A Pb–Ti–Zr complex alkoxide was synthesized by mixing lead acetate trihydrate [Pb(OCOCH3)2·3H2O] (≥ 99%, Aldrich), zirconium (IV) butoxide [Zr(OC4H9)4] (80 mass% in 1-butanol, Aldrich), and titanium (IV) isopropoxide [Ti(OC3H7)4] (97%, Aldrich) in isopropanol [(CH3)2CHOH] (99.5%, Sigma-Aldrich) under nitrogen gas flow (N2), and the solutions were protected from moisture. The metal ions were solubilized through the formation of complexes with DEA [(HOCH2CH2)2NH] (99%, Aldrich) (PZT-DEA precursor) or acetoin [CH3COCH(OH)CH3] (Aldrich) (PZT-AC precursor), which were added under a constant stirring. This procedure ensures a homogeneous mixing at a molecular level of all metal cations, thereby favoring the crystallization by reducing the diffusion path range.
The gel powders were then thermally treated in an open-end crucible and inserted into a tube furnace previously set at the appropriate temperature at a heating rate of 5 °C min−1. At the end of the desired treatment time, the powders were removed and cooled in air. The heat treatments were accomplished under permanent flow of air or pure oxygen through the furnace.
The gel powders’ thermal decomposition was monitored by simultaneous thermogravimetric–differential thermal analysis (TG–DTA). TGA/DTA was recorded on a thermal analyzer (Model TA-50 WS, Shimadzu, Kyoto, Japan) under flowing air. The heating rate varied between 7.5 and 17.5 °C min−1. The crucial thermal transformations occurring in the temperature range RT-800 °C were identified. Kissinger’s method [7] and JMAK approach were applied to evaluate the energy barrier for the crystallization of PZT perovskite phase using DTA. The samples were heat-treated from room temperature to 800 °C at different heating rates. The temperatures of the DTA exothermic peaks, which correspond to the perovskite phase crystallization from both systems, were determined. The X-ray diffractograms were recorded at room temperature on a Rigaku diffractometer (Model RAD-2R, Rigaku Co., Tokyo, Japan) in reflection mode with CuKα radiation (40 kV, 30 mA). The pyrochlore and perovskite phases were quantitatively analyzed using Origin software.
Theoretical background: determination of the crystallization kinetic parameters by non-isothermal phase transformation
Kissinger approximation
The Kissinger method is often used to evaluate the kinetic parameters of the crystallization through the interpretation of DTA results. The method was developed by Kissinger [7, 8] and further modified by Starink [9] for a more precise evaluation of the activation energy.
According to the Kissinger’s formula,
where \( \emptyset \) refers to the heating rate, \( E_{\text{a}} \) is the activation energy, R is the universal gas constant, and \( T_{\text{m}} \) is demarcated as the peak temperature.
The crystallization rate factor can then be calculated using the Arrhenius equation:
here \( K_{\text{o }} \) is the pre-exponential (or frequency) factor.
The activation energies corresponding to the crystallization of the perovskite phase can be estimated from the slope of the plot of \( \left( {\ln \emptyset /T_{\text{m}}^{2} } \right) \) versus \( \left( {1/T_{\text{m}} } \right) \). The consistence of the activation energy values calculated by this model may be limited since Ea is assumed to be constant during the reaction. Nevertheless, the Kissinger approach can still provide a basic comparative idea of the crystallization kinetic data of a certain compound prepared via different routes. However, the results should not be considered as absolutely valid.
The pre-exponential factor can be deduced from the intercept following:
The knowledge of the crystallization rate factor \( K_{\text{T}} \) can thus be calculated according to Eq. 2. The experimental procedure includes a number of DTA runs of the PZT gel powder at diverse heating rates \( \emptyset \).
Johnson–Mehl–Avrami–Kolmogorov (JMAK) approach
The kinetics of isothermal phase transformation arising by nucleation and growth can be described by the JMAK model. The JMAK approach relates the crystallized fraction \( \alpha \left( t \right) \) to time (t) in agreement with the transformation kinetic equation:
where k is the crystallization rate constant (which varies the nucleation frequency and growth rate but differs from \( K_{\text{T}} \) in the reaction kinetics), and \( n \) is the exponent which does not depend on temperature but sensitive to the reaction mechanism. k as well as n can be resoluted by fitting Eq. 4.
Equation 4 can be rearranged in a similar form:
The kinetic exponent n keeps the same value. According to the JMA model, the isothermal crystallization rate can be found by differentiating Eq. 5 with respect to time. The resulting expression has the following form:
Several research groups have reported [10,11,12,13,14] that the validity of the JMA theory can be extrapolated to the analysis of non-isothermal crystallization, provided that:
-
1.
The crystals phase grows from a constant number of nuclei.
-
2.
Nucleation was completely achieved prior to the crystal growth.
When the nucleation phenomenon entirely occurs in the course of early stage of the transformation, the crystallization is referred to as isokinetic process and its rate depends only on temperature. Under these conditions, Eq. 6 can be applied to non-isothermal circumstance.
The temperature dependence of the kinetic constant k appearing in Eqs. 5 and 6 has a simple Arrhenius comportment during the crystallization:
here the constant A is the pre-exponential factor. Under the above-mentioned conditions, the rate equation describing a non-isothermal process can be estimated by combining Eqs. 6 and 7:
Taking the logarithm of the integrated Eq. 10, the subsequent equation is obtained [15]:
where \( x = \frac{{E_{\text{a}} }}{RT} \) and \( \pi \left( x \right) = \frac{{x^{3} + 18x^{2} + 88x + 96}}{{x^{4} + 20x^{3} + 120x^{2} + 240x + 120}} \) (Ref. [16]).Considering the term \( \ln \left[ {\frac{AT}{\emptyset }\pi \left( x \right)} \right]^{\text{n}} \) as a constant, a plot of \( \ln \left[ { - \ln \left( {1 - \alpha } \right)} \right] \) versus the reciprocal temperature \( \frac{1}{T} \) should be linear, having a slope equal to \( - \frac{{nE_{\text{a}} }}{R} \) [15]. However, this linear relationship does not systematically imply the applicability of the JMA model to interpret any thermal experimental data. In fact, as pointed out by Málek [15], this is due to the low sensitivity of the double logarithmic function as well as the temperature dependence of the term \( \ln \left[ {\frac{AT}{\emptyset }\pi \left( x \right)} \right]^{\text{n}} \), which cannot always be neglected. When the aforementioned conditions are fulfilled, the JMAK approach can also be used to determine the Avrami exponent using Ozawa’s equation for linear heating rates [13]:
The value of α is the ratio of the partial integrated area to the perovskite crystallization peak at a fixed temperature over the total peak integration area.
Microstructure evolution
The main issue in the crystallization of lead zirconate titanate is the formation of an intermediate fluorate- or pyrochlore-phase Pb2Ti2O7−x with 0 ≤ x ≤ 1. Pb2Ti2O7−x is a lead-deficient phase with cubic symmetry and as a consequence lacks ferroelectric properties. Despite the stability of the perovskite form, the development of the metastable fluorate or pyrochlore is kinetically favored since the titanium neighborhood of the amorphous state largely resembles that of Pb2Ti2O7−x [17, 18]. Several alternatives have been proposed to diminish the content of this undesired phase in the perovskite. The most widely used approaches are the inclusion of lead excess in the starting precursor system and/or the heat treatment of the powders under PbO-saturated atmosphere. Several researchers have pointed out the importance of the processing parameters on the crystallization pathway either through the lead-deficient transient phase or directly to the perovskite form. Completely avoiding pyrochlore phase remains a complex task. Two-stage crystallization with the pyrochlore as the intermediate phase often takes place.
Intermediate phases
PZT-DEA system
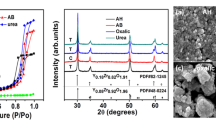
The gel powders were heat-treated quasi-isothermally in a preheated tube furnace at different temperatures and durations. The intermediate phases were identified by analyzing the XRD signals. Basically, two crystalline phases evolved before perovskite formation. At first, Pb reflections emerged at temperatures higher than 300 °C, followed by PbO signals that started at around 400 °C. Both phases coexisted up to 450 °C. The typical spectra shown in Fig. 1 depict this situation.

X-ray diffraction patterns for powders prepared from PZT-DEA precursor and heat-treated under air at 5 °C min−1 up to the temperatures indicated and then quenched to room temperature. The spectra are shifted along the ordinate for lucidity
At the first combustion stage, lead was reduced to zero valent before being oxidized. The decomposition of organic materials provided stable reducing conditions. The same situation has been previously observed for PZT gel with the high level of residual carbon [19]. Elemental Pb is crystallized according to the following reaction:
Several authors attributed the formation of elementary Pb to the reduction of lead oxide by organic mass [20]. This is not likely to happen in the present work since the PbO was formed through the oxidation of Pb which has been crystallized earlier.
The lead re-oxidation apparently starts after their complete combustion of the organic mass. When increasing the calcining temperature or extending the heat-treatment dwell time, the XRD signals of lead oxide progressively diminished at the expense of perovskite. PZT perovskite phase started to form at 400 °C. However, pure PZT phase could not be obtained at this temperature even after extending the calcining duration up to 10 h (Fig. 2). The crystallization of the perovskite phase at low temperature will be sufficient in reducing the major part of Pb loss due to volatilization.

Room-temperature powder X-ray diffractograms of the PZT powders from PZT-DEA precursor after heat treatment in air: a for 3 h and b for 10 h at the temperatures indicated
Compared with other reports, the preparation scheme developed in this study has efficiently decreased the crystallization temperature of single perovskite PZT phase to 450 °C. As will be seen later, this result is consistent with the remarkably low activation energy relative to the PZT formation.
The combustion of the organic mass is highly exothermic, and thereby, considerable thermal energy is provided which favors the perovskite formation under the moderate external temperature. Fukui [21] considered that extended reaction time is essential to increase the perovskite level at a reasonable temperature. Schwartz et al. [22] attributed the low crystallization temperature of powders synthesized via chemical routes to relatively low activation energy. The formation of the intermediate pyrochlore phase should not also be ignored. The following paragraphs will closely examine to which extent these points influence the crystallization behavior.
PZT-AC system
The thermal transformation of the gel processed from this system followed different paths to the perovskite formation. Figure 3 depicts the corresponding XRD spectra. The perovskite phase initiated concomitantly with pyrochlore at T = 400 °C. The intensity of the Pyrochlore reflections starts diminishing as the calcining time increases. Extended calcination time up to 10 h was insufficient to totally covert the pyrochlore-to-perovskite phase. The perovskite phase predominated with further increase in the firing temperature and time as evidenced by the sharpening of the correspondent peaks. Single phase could be obtained at 650 °C/3 h. The nucleation and growth of the perovskite phase from the intermediate pyrochlore require a certain minimum Pb level [23]. Therefore, in the present case, the perovskite likely formed through the subsequent diffusion of Pb into the pyrochlore.

X-ray diffraction patterns for powders prepared from PZT-AC precursor and heat-treated in air at 5 °C min−1 up to the temperatures indicated for a 0 h, b 10 h, c and d 3 h then quenched to room temperature
The same experiments have been conducted under flowing oxygen to determine the impact of the calcining atmosphere on the gel pyrolysis. Figure 4 shows the X-ray spectra along with that of the as-dried gel. The presence of broad humps around 2θ = 29 and 35° indicates early-stage crystallization of pyrochlore structure which continues at higher temperatures as evinced by the appearance of the (311) and (400) reflections. The utilization of pure oxygen atmosphere seems to hamper and retard the crystallization of the PZT perovskite phase. Pure oxygen probably promoted the crystallization of pyrochlore phase by increasing the number of nucleation sites. Compared with the pyrochlore phase, the perovskite is known to be relatively enriched with lead and deficient in Zr and oxygen. Therefore, the formation of metallic lead, when the PZT-DEA system has been used, may have helped the direct crystallization of the PZT perovskite from the amorphous phase.

X-ray diffraction patterns for powders prepared from PZT-AC precursor and a heat-treated under oxygen at 5 °C min−1 up to the temperatures indicated and then quenched to room temperature, and b for 3 h
Kinetic parameters
Crystallization kinetics derived from Kissinger approximation
The results of the DTA are analyzed after partial solvent evaporation. The DTA curves recorded at different heating rates for PZT samples from the PZT-AC system are shown in Fig. 5. The DTA peaks broadened and shifted to higher temperatures as the heating rate is getting larger. The atomic rearrangement was postponed to higher temperature as the heating rate increased since the decomposition depends on heat-transfer time. The peak broadening indicates enhanced crystal growth. The development of the crystalline perovskite fraction as a function of temperature (time) from PZT-AC gel at various heating rates is shown in Fig. 6. The sigmoidal curves seen in this figure are typical for a crystallization progression comprising of an initial period of induction followed by fast crystallization of the perovskite phase and regular growth of the crystallites until a constant crystallite size value. The temperature relative to a fixed crystallized fraction increases as the heating rate increases, due to shorter conversion time. The activation energy and pre-exponential factor of crystallization are calculated in Eqs. 1–3. The linear regression of Kissinger’s formula (Eq. 1) for different heating rates is shown in Fig. 7 for the PZT-AC and PZT-DEA systems. Even though the analysis was limited to the PZT crystallization, the Kissinger approach was not applicable to certain DTA data that have been recorded at particular heating rates due to the overlapping with various other decomposition steps. Figure 8 illustrates an example of DTA peak that could be deconvoluted into two distinct peaks. The DTA curves as well as the TGA analysis presented in Fig. 9 revealed that the gels derived from both precursors decomposed their organic contents at comparable temperatures. When the DTA peak records were interpreted by means of the Kissinger approach, it became evident that comparable combustion temperatures were related to analogous activation energies. In addition, the decomposition of PZT-AC- and PZT-DEA-type precursor gels proceeded at similar rates once the energy barrier was overcome as the resultant pre-exponential factors showed (Table 1). The accordant results are pictured in Fig. 9. The apparent activation energies calculated in the case of the present work are numerically summarized in Table 1. The activation energies reported for other sol–gel systems are shown in Table 2. The formation of the crystalline perovskite lattice from both systems consumed reasonably low activation energy and progressed at a moderate rate. The kinetics of the crystallization of PbTiO3 powders from metallo-organic precursors has been previously described by Shaikh [24], and activation energy values within the range of 60–87 kJ mol−1 were reported.

Representative DTA curves for PZT gel powders from PZT-AC system with different heating rates

Development of the perovskite fraction as a function of temperature for various heating rates corresponding to PZT-AC

Kissinger’s plots corresponding to the crystallization of PZT perovskite phase from a PZT-AC and b PZT-DEA precursor systems

(Color online) Exothermic peaks of DTA curve recorded at heating rate of 12.5 °C min−1 for PZT gel powder synthesized from PZT-DEA precursor. (1) Experimental curve, (2) Gaussian fit of cumulative peak, and (3) and (4) are the deconvoluted peaks

DTA/TGA curves for PZT gel powders from PZT-AC and PZT-DEA systems recorded at a heating rate of 7.5 °C min−1
Based on the activation energy values calculated for pyrochlore-to-perovskite phase transformation, Chang et al. [25] deduced that the pyrolysis time defines the formation mechanism (nucleation or growth-controlled) of the perovskite form. They attributed the relatively low activation energy to the formation of crystalline PbO that serves as seeding sites for PZT crystals. Polli and Lange [20] followed three different methods to prepare PZT powders. They found that the carbonaceous content in the gel and the rate of hydrocarbon escape during pyrolysis are essential to prevent the formation of elemental Pb. Durán et al. [26] as well as Chen et al. [27] attributed the difference in the estimated activation energies to the various precursor compounds and manufacturing processes that result in T poly-crystals. This interpretation seems not to apply in the present case since different crystallization paths and precursors solutions led to comparable activation energies. James et al. [28] related this discrepancy to the difference in the source of metal precursor as well as other reaction conditions.
The activation energy should account for the energy required for complex destabilization and PZT crystallization. Since the crystallographic structure of the pyrochlore from which the perovskite phase initiated was the same, the discrepancy in the reported activation energies partly resulted from the difference in the complex destabilization energy. In addition, the homogeneous mixing of the cations at molecular level reduced the metallic diffusion path and thereby facilitated the crystal formation.
Crystallization kinetics derived from the JMAK approach
Experiments were undertaken in order to evaluate kinetic parameters from non-isothermal (DTA) procedure through the application of the JMAK approach. Non-isothermal conditions during DTA measurements were realized in the DTA/TGA apparatus chamber. The obtained results were analyzed by applying the original approach [Eq. (6)] and the derivative equation for non-isothermal experimentations (Eqs. 9 and 10) to the DTA measurement results.
The exponent n is deduced from the slope of the fitted linear relationship of \( \ln \left[ { - \ln \left( {1 - \alpha } \right)} \right] \) versus \( {\text{Ln}}\emptyset \) at various temperatures, as shown in Figs. 10 and 11. The obtained exponent n for the exothermic peak relative to the crystallization of PZT perovskite form from both precursor systems is collected in Table 3. The exponent value (3.5) obtained for the PZT powder prepared from the PZT-AC system indicates that the crystallization mechanism relates to an increasing (constant) nucleation rate with diffusion-controlled growth. Figure 12 shows the plot used to calculate the activation energy for the crystallization of PZT from the PZT-AC system. The plots are reasonably straight in the selected intervals. The slope of \( { \ln }\left[ { - { \ln }\left( {1 - \alpha } \right)} \right] \) against 1/T yields activation energy of 77 kJ mol−1, which is close to that obtained using Kissinger’s model. This indicates that both approaches are applicable to analyze the crystallization of PZT from this system. Therefore, the JMAK model can be extended to analyze the crystallization under non-isothermal circumstances when the conditions mentioned in the earlier paragraph are satisfied.

Plots of ln[− ln (1 − α)] against ln (\( \emptyset \)) for non-isothermal crystallization of PZT powder from PZT-AC system analyzed according to the JMAK model at various temperatures

Plots of ln[− ln (1 − α)] against ln(\( \emptyset \)) for non-isothermal crystallization of PZT powder from PZT-DEA system analyzed according to the JMAK model at various temperatures

JMAK plots of ln[− ln (1 − α)] as a function of the reciprocal temperature for the crystallization of PZT from PZT-AC system
Example of the plot \( { \ln }\left[ { - { \ln }\left( {1 - \alpha } \right)} \right] \) versus 1/T corresponding to the PZT perovskite fraction from the PZT-DEA system is depicted in Fig. 13. The Avrami exponent as well as the activation energy is collected in Table 3. The Kissinger evaluation and the JMAK analysis yielded approximately equivalent values concerning the activation energies associated with crystallization. The Avrami exponents of 2.6 and 3.8 indicate a three-dimensional growth with a crystallization occurring at a constant nucleation rate.

JMAK plots of ln[− ln (1 − α)] as a function of the reciprocal temperature for the crystallization of PZT from PZT-DEA system
Applicability of the JMAK model
As previously mentioned in “Johnson–Mehl–Avrami–Kolmogorov (JMAK) approach” section, the perfect linearity of the double logarithm versus the reciprocal temperature should not be always taken as an absolute argument to justify the validity of the JMAK theoretical model. Although the rigorous and trustworthy test is the direct comparison of the experimentally measured data with the theoretical curve, several authors have suggested reliable tests to validate the applicability of the JMAK approach.
Málek [15] proposed another test based on \( Z\left( \alpha \right) \) function which is defined by:
This maximum function should belong to the interval 0.62 < \( \alpha_{\text{M}} \) < 0.64. Figures 14 and 15 show that the maximum value of the function \( Z\left( \alpha \right) \) is broad and falls slightly outside this interval. The curves shown in Figs. 14 and 15 have the same shape as that of the theoretical function (Refs. [13, 15]). Málek [15] attributed the difference from theoretical value (0.632) to an approximation of the \( \pi \left( x \right) \) function (Eq. 9). Based on Henderson’s testing method [11], the same author considered that \( \alpha_{\text{M}} \) values near 0.632 could be wrongly interpreted to mean that the results do not agree with the JMA approach. Therefore, the slight deviation from the theoretical values does not necessarily exclude the validity of the JMAK analysis.

Normalized Z(α) and derivative (α) curves calculated for the JMA model (precursor system: PZT-AC, β = 10 °C min−1

Normalized Z(α) and derivative (α) curves calculated for the JMA model (precursor system: PZT-DEA, β = 7.5 °C min−1
Conclusions
The perovskite formation followed different routes depending on the precursor system. XRD analysis did not reveal any signal of the metastable pyrochlore phase when PZT-DEA precursor system has been used. In contrast, the nanocrystalline pyrochlore phase mediated the perovskite which has been formed from PZT-AC solution. The composition stability throughout the PZT powder processing is provided for good reproducibility, and favored the crystallization of the PZT perovskite form at a relatively low temperature of 400 °C compared to other reports. This result is consistent with the low activation energies corresponding to the PZT formation. The XRD spectra showed an incessant, regular upturn of the conversion ratio of the perovskite phase with the increase in calcination time and temperature. The utilization of pure oxygen atmosphere during the gel pyrolysis did not minimize the intermediate phase content as expected. In contrast, nanocrystals of the transient pyrochlore phase, which is more stable at low temperatures, were developed.
Kissinger and JMAK models have been used under non-isothermal circumstances to analyze the PZT perovskite crystallization kinetics with morphotropic composition. The apparent activation energy estimated from the Kissinger model was about 75 kJ mol−1 regardless of the precursor system. Nearly the same value was calculated using the JMAK approach for PZT from PZT-AC system, whereas higher activation energy was obtained for PZT from PZT-DEA system applying the same model. The activation energies found in this work remain among the lowest reported. The transformation into the perovskite form has an Avrami exponent of about 3, which can be interpreted by saturated nucleation followed by three-dimensional growth.
References
Huang H, An Z, Huang P, Li D, Liao Q. Phase transition and thermal deposition of double perovskite modified low sintering temperature PbTiO3 piezoelectric ceramics. J Eur Ceram Soc. 2016;36:925–9.
Muralt P. Texture control and seeded nucleation of nanosize structures of ferroelectric thin films. J Appl Phys. 2006;100:051605.
Fernandes JC, Hall DA, Greaves GN. Crystallization of gel-processed PZT powders. Mater Sci Forum. 1996;228–231:411–6.
Merkle R, Bertagnolli H. Investigation of the crystallization of lead titanate and lead zirconate titanate with X-ray diffraction and differential thermal analysis. Ber Bunsenges Phys Chem. 1998;102:1023–31.
Bel-Hadj-Tahar R, Bel-Hadj-Tahar N, Salah AB. Low-temperature processing and characterization of single-phase PZT powders by sol–gel method. J Mater Sci. 2007;42:9801–6.
Bel-Hadj-Tahar R, Bel-Hadj-Tahar N, Salah AB. Preparation and characterization of PZT solid solutions via sol–gel process. J Cryst Growth. 2007;42:9801–6.
Kissinger HE. Reaction kinetics in differential thermal analysis. Anal Chem. 1957;29:1702–6.
Kissinger HE. Variation of peak temperature with heating rate in differential thermal analysis. J Res NBS. 1956;57:217–21.
Starink MJ. The determination of activation energy from linear heating rate experiments: a comparison of the accuracy of isoconversion methods. Thermochim Acta. 2003;404:163–76.
Henderson DW. Experimental analysis of non-isothermal transformations involving nucleation and growth. J Therm Anal. 1979;15:325.
Henderson DW. Thermal analysis of non-isothermal crystallization kinetics in glass forming liquids. J Non Cryst Solids. 1979;30:301.
DeBruijn TJW, DeJong WA, Van Den Berg PJ. Kinetic parameters in Erofeev type reactions from isothermal and non-isothermal experiments. Thermochim Acta. 1981;45:315.
Málek J, Mitsuhashi T. Testing method for the Johnson–Mehl–Avrami equation in kinetic analysis of crystallization processes. J Am Ceram Soc. 2000;83:2103–5.
Perez-Maqueda LA, Criado JM, Malek J. Combined kinetic analysis for crystallization kinetics of noncrystalline solids. J Non Cryst Solids. 2003;320:84–91.
Málek J. The applicability of Johnson–Mehl–Avrami model in the thermal analysis of the crystallization kinetics of glasses. Thermochim Acta. 1995;267:61–73.
Šesták J. Thermophysical properties of solids, their measurements and theoretical analysis. Amsterdam: Elsevier; 1984.
Feth MP, Weber A, Merkle R, Reinohl U, Bertagnolli H. Investigation of the crystallization behavior of lead titanate (PT), lead zirconate (PZ) and lead zirconate titanate (PZT) by EXAFS spectroscopy and X-ray diffraction. J Sol-Gel Sci Technol. 2003;27:193–204.
Camargo ER, Longo E, Leite ER, Mastelaro VR. Phase evolution of lead titanate from its amorphous precursor synthesized by the OPM wet-chemical route. J Solid State Chem. 2004;177:1994–2001.
Wilkinson AP, Speck JS, Cheetham AK, Natarajan S, Thomas JM. In situ x-ray diffraction study of crystallization kinetics in PbZr1−xTixO3, (PZT, x = 0.0, 0.55, 1.0). Chem Mater. 1994;6:750.
Polli AD, Lange FF. Pyrolysis of Pb(Zr0.5Ti0.5)O3 precursors: avoiding lead partitioning. J Am Ceram Soc. 1995;78:3401–4.
Fukui T. Influence of preparation conditions on crystallization behavior of the complex alkoxide derived PbTiO3 Powder. J Sol-Gel Sci Technol. 1998;11:31–41.
Schwartz RW, Payne DA. Crystallization behavior of chemically prepared and rapidly solidified PbTiO3. In: Brinker CJ, Clark DE, Ulrich DR, editors. Better ceramics through chemistry III. Pittsburgh: Materials Research Society; 1988. p. 199–206.
Bose A, Sreemany M, Bysakh S. Role of TiO2 seed layer thickness on the nanostructure evolution and phase transformation behavior of sputtered PZT thin films during post-deposition air-annealing. J Am Ceram Soc. 2011;94:4066–77.
Shaikh A, Vest G. Kinetics of BaTiO3 and PbTiO3 formation from metallo-organic precursors. J Am Ceram Soc. 1986;57:682.
Chang TI, Wang SC, Liu C, Lin CF, Huang JL. Thermal behaviors and phase evolution of lead zirconate titanate prepared by sol–gel processing: the role of the pyrolysis time before calcination. J Am Ceram Soc. 2008;91:2545–52.
Duran P, Tartaj J, Fernandez JF, Villegas M, Moure C. Crystallisation and sintering behaviour of nanocrystalline Y-TZP powders obtained by seeding-assisted chemical coprecipitation. Ceram Int. 1999;25:125.
Chen YF, Wang MC, Hon MH. Phase transformation and growth of mullite in kaolin ceramics. J Eur Ceram Soc. 2004;24:2389.
Eckert JO Jr, Hung-Houston CC, Gersten BL, Lencka MM, Riman RE. Kinetics and mechanisms of hydrothermal synthesis of barium titanate. J Am Ceram Soc. 1996;79:2929–39.
Khajelakzay M, Taheri-Nassaj E. Crystallization kinetics and growth mechanism of Pb(Zr0.52Ti0.48)O3 nanopowders. Appl Phys A. 2014;116:179–83.
Babushkin O, Lindbäck T, Brooks K, Setter N. PZT phase-formation monitored by high-temperature X-ray diffractometer. J Eur Ceram Soc. 1997;17:813.
Chang TI, Huanga JL, Lin HP, Wang SC, Lu HH, Wu L, Lin JF. Effect of drying temperature on structure, phase transformation of sol–gel-derived lead zirconate titanate powders. J Alloys Compd. 2006;414:224–9.
Acknowledgements
This work was supported by the Deanship of Scientific Research, King Khalid University, Abha, KSA [Grant Number 432-39, 2018].
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bel-Hadj-Tahar, R., Abboud, M. & Bouzitoun, M. Thermal analysis of the crystallization kinetics of lead zirconate titanate powders prepared via sol–gel route. J Therm Anal Calorim 144, 127–138 (2021). https://doi.org/10.1007/s10973-020-09439-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-020-09439-8