
Abstract
Polypropylene block copolymer (PPco) is easier to process in extrusion-based additive manufacturing compared to isotactic PP homopolymer because it shrinks and warps less during printing. This study investigated the thermal properties of PPco and spray-dried CNF (SDCNF)-PPco composite pellet formulations. Thermogravimetric analysis data showed that the addition of SDCNF lowered the thermal degradation onset temperature and increased the residual mass content (at 450 °C) of PPco pellets. The peak degradation temperatures of SDCNF and PPco remained the same. The peak crystallization temperature and relative crystallinity of PPco were increased by the addition of SDCNF and MAPP. The peak melting temperature of PPco was not significantly changed. Printed specimens showed three melting peaks (β, β′ and α crystals) while injection molded PPco only showed one (α crystal) melting peak. Dynamic mechanical analysis results showed that the heat deflection temperatures of printed SDCNF-PPco composites were higher than injection molded PPco. SEM micrographs showed that the addition of SDCNF increased the number of voids inside the printed parts.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Extrusion-based additive manufacturing (AM) has been used to print thermoplastics and their composites [1]. Other names for extrusion-based AM include fused deposition modeling (FDM), fused layer modeling (FLM), fused filament fabrication (FFF) and material extrusion [2]. Because of its low cost and simple operation, extrusion-based AM is being extensively investigated [3]. However, extrusion-based AM is not suitable for printing isotactic PP homopolymer (iPP) because it warps significantly as compared to other popular thermoplastics used in AM, for example, Poly(l-lactic acid) (PLLA). The rapid crystallization rate is the major factor contributing to iPP’s warping. Impact-modified polypropylene, or polypropylene block copolymer (PPco), was reported to crystallize more slowly than iPP [4]. Therefore, PPco can be printed more easily. Figure 1 shows the comparison of the 3D printing processability of iPP and PPco.

IPP (back) and PPco (front) specimens from 3D printing
Cellulose nanofibers typically refer to cellulose nanofibrils (CNF), cellulose nanocrystals (CNC) and bacterial cellulose (BC) [5]. CNF is produced through mechanical fibrillation of pulp fibers and possesses a diameter on the nanoscale and lengths on the micron scale [6]. CNC is obtained via acid hydrolysis of pulp whose diameter and length are both on the nanoscale. Because of its high aspect ratio, CNF was reported to outperform CNC in reinforcing various polymers [7]. To facilitate the compounding of CNF with thermoplastics, dried CNF is desired [8]. Spray-dried CNF (SDCNF) has been demonstrated to be an effective way to obtain dried powder [8].
Thermal analysis of natural fiber-filled PP composites has been reported widely [9,10,11,12,13]. Cellulose nanofibers are distinct from natural fibers based on their chemical composition, surface properties and fiber morphology. CNF and CNC were reported to increase the peak crystallization temperature (Tc) of PP [14, 15]. In other studies, CNC increased the crystallinity (Xc). The peak melting temperature (Tm), Tc and residual mass of PP were unchanged [16, 17]. However, CNC or CNF was also reported to decrease the Xc of PP [14, 18]. Cellulose nanofibers reduced the onset temperature (To) of PP composites because they are less thermally stable than PP [19, 20]. Meanwhile cellulose nanofibers retarded the thermal degradation mass loss of PP at high temperatures [19]. Cellulose nanofibers did not affect the Xc of PP. A coupling agent, maleic anhydride polypropylene (MAPP), is often used in making cellulose nanofiber-filled PP composites. However, the influences of MAPP on thermal properties (Tm, Tc, Xc) vary among reported research most likely attributable to chemical makeup of the coupling agent [14, 15, 17, 18].
Thermal-related process settings (extrusion and bed temperatures) used in commercial 3D printing software are often recommended based on qualitative experience. It is important to understand precisely when materials melt and crystallize and select the right 3D printing parameters to reduce materials’ thermal degradation and improve objects’ material properties. Moreover, heat deflection temperature of 3D-printed cellulose-PP composites has not been explored. In this study, thermal analysis was performed to better understand the processing of SDCNF-PPco composites in extrusion-based AM. Thermogravimetric analysis (TGA) was used to obtain the To of such composites to avoid thermal degradation during printing. Differential scanning calorimetry (DSC) was utilized to determine the Tm and Tc of SDCNF-PPco pellets to help choose proper printing and build bed process temperatures. DSC was also performed on manufactured SDCNF-PPco parts to obtain Xc. Dynamic mechanical analysis (DMA) was conducted to measure heat deflection temperature (HDT) of the processed composites. HDTs of printed PPco and SDCNF-PPco composites were compared with injection molded PPco. Scanning electron microscopy (SEM) was used to visualize the inner morphological structure of the printed parts.
Materials and methods
Materials
CNF suspension (~ 3 mass%) was produced by the Product Development Center at the University of Maine via a mechanical grinding process. This original CNF suspension was diluted to 1.2 mass% solids content for spray drying. A pilot-scale spray dryer (GEA-Niro, Germany) was used to dry the CNF suspension at a chamber temperature of 250 °C, pump feeding rate of 0.4 L min−1 and a disk spinning rate of 30,000 rpm. The mean diameter of SDCNF is around 10 µm. The aspect ratio of SDCNF is 1.25. The polypropylene impact copolymer (ExxonMobil TM PP7414) formulated for automotive applications was purchased from ExxonMobil (Texas, USA). The PPco had a melt flow index (MFI) of 20 g 10 min−1, a density of 0.9 g cm−3, an Izod impact strength of 180 J m−1 and a HDT of 85.1 °C. MAPP pellets (Polybond 3200) were purchased from Chemtura Corporation (Lawrenceville, GA). It has a MA content of 1.0 mass%, a density of 0.91 g cm−3 and a MFI of 115 g 10 min−1.
Compounding
To improve the distribution and dispersion of SDCNF within the PPco, a masterbatch compounding process was used. The detailed description of this process can be found in a previous paper [2]. PPco, MAPP and SDCNF were conditioned at 105 °C for at least 2 h. PPco, MAPP and SDCNF (30 mass%) were hand mixed and fed into a corotating twin screw extruder (C.W. Brabender Instruments, South Hackensack, NJ) at an approximate feeding rate of 7 g min−1, a screw speed of 200 rpm and a temperature of 200 °C for all five heating zones. Extrudates were granulated using a grinder (Hellweg MDS 120/160, Hackensack, NJ). During a second extrusion step, fresh PPco was added to the masterbatch to dilute it to the final composite formulation mass percentages which are listed in Table 1. Feeding rate, screw speed and heating zone temperatures for the second extrusion step were maintained comparable to the first extrusion. Extrudates were again granulated. Composite granules were fed into the extruder outfitted with a nozzle die (2.7 mm) to produce filaments for AM during the third extrusion. The feeding rate was about 3 g min−1, screw speed was 60 rpm, five-heating zone profile was 185 °C, 180 °C, 175 °C, 172 °C, 170 °C, and the die temperature was 165 °C. After the die, the composite extrudate passed through a water tank (room temperature) to solidify the shape of filament and was wound on spools.
Composite part manufacturing
Composites and pure PPco parts were printed using a LulzBot TAZ 6 (Aleph Objects Inc., Colorado, USA) with Cura printing software (Version 21.03). All specimens were printed one at a time at the same location on the printing bed. To adhere the extruded PPco strands to the bed, a strip of PP-based packing tape (Office Depot®, OfficeMax #24767995) was adhered on top of the bed. Important AM printing settings were extrusion temperature of 200 °C, bed temperature of 120 °C, extrusion speed of 45 mm s−1, infill density of 100%, layer height of 0.3 mm, number of shell of 2, with brim, orientation of ± 45° and airspace of 0%. Only pure PPco pellets obtained from the PPco filament were injection molded and used as a control specimen (PPcoIM). Injection molding was done on a “Minijector” (Model #50) at a temperature of 200 °C, a pressure of 17 MPa and a holding time of 10 s with a room temperature mold. Additional composite filaments were also chopped into pellets for thermal analysis.
Thermogravimetric analysis (TGA)
TGA measurements on PPco and SDCNF-PPco composites pellets were conducted using a TA Instruments Q500 (New Castle, Delaware, USA). About 5–10 mg samples were used for each measurement. Samples were heated from 30 to 450 °C at a heating rate of 10 °C min−1 under nitrogen to avoid oxidation. Derivative thermogravimetric (DTG) analysis results were obtained simultaneously. At least, three individual samples for each formulation were randomly picked from the granules for testing.
Differential scanning calorimetry (DSC)
The DSC was performed on both pellets and printed samples using a TA Instruments Q 2000 (New Castle, Delaware, USA). In each case, a piece of 1–3 mg was cut from the larger sample, placed in a pan and sealed. For pellets, they were heated to 190 °C at a rate of 10 °C min−1 and held at that temperature for 5 min to erase thermal history before being cooled down to 30 °C. Tc was obtained during the cooling process. Then samples were again heated up to 190 °C to obtain Tm and Xc. For printed samples, they were directly heated to 190 °C at a rate of 10 °C min−1. The Xc of composite pellets and injection molded PPco were calculated from the following equation [3],
where ΔHm is melting enthalpy of PP, ∆H 0f is the fusion enthalpies of PP which displays 100% crystallinity and was reported to be 209 J g−1 from the literature [3]. The ϕ is the percentage of the polymer in the composites. Because no information regarding the percentage of rubber phase in the PPco was provided by the polymer provider, we considered the percentage of PP inside PPco as 100% and named the Xc as relative crystallinity (RXc). Three replicates were tested for each sample.
DMA
DMA was used to obtain the HDT of PPco and its printed composites using the TA Instruments Q 800 (New Castle, Delaware, USA). A dual-cantilever mode was used for the testing which was performed according to ASTM D 648. A constant force was exerted on the sample bar which later experienced a temperature change from 40 to 120 °C at a heating rate of 2 °C min−1. The HDT was defined as the temperature where the test bar deflects by 0.25 mm. The constant force can be calculated from the following equation,
where σ is the maximum stress (0.455 MPa), T is thickness, W is width, and L is length (35 mm) of the sample. Three replicates were measured for each specimen.
Scanning electron microscopy (SEM)
SEM (TM 3000, Hitachi High-Technologies Corporation, Tokyo, Japan) was used to observe the morphological structure of specimens obtained from injection molding and AM. The testing was done on impact-fractured surfaces at an accelerating voltage of 5 kV.
Results and discussion
Thermal gravimetric analysis (TGA)
The TGA and DTG graphs of SDCNF, PPco and SDCNF-PPco composites are shown in Fig. 2. Because all SDCNF-PPco composites displayed similar graphs, only the PPcoMASDCNF15 sample was shown for simplicity purposes. Important information extracted from TGA and DTG curves are listed in Table 2. The To of SDCNF is lower than PPco. Therefore, once SDCNF was added into PPco, the To of the composites decreased compared to PPco. The TDTG-SDCNF remains similar between SDCNF and SDCNF-PPco composites. The TDTG-PPco remains close between PPco and SDCNF-PPco composites. These findings are consistent with a previous study on the thermal stability of CNF reinforced PPco [19]. This is because the degradation peak temperature of SDCNF or PPco, as an intrinsic property, is not affected by the compounding process. As seen in Fig. 2b, adding SDCNF into PPco reduces the peak degradation rate (derivative mass) of SDCNF because of the embedding of SDCNF in PPco matrix, while the existence of SDCNF hardly impacted the peak degradation rate of PPco. This indicates that once the SDCNF starts degrading, it decomposes more slowly in the PPco matrix than in the nitrogen atmosphere. This result was also found in a previous study [19]. The TDTG-PPco in our work was ~ 380 °C which is lower than a reported value (~ 430 °C) [19]. The PPco used here went through three extrusion processes where two of them were performed at high screw speed (200 rpm). Multiple extrusions, especially at high screw speed, are reported to cause chain scission and molecular weight reduction. Those changes make PP less thermally stable [21, 22]. Lower mechanical properties (not shown) obtained from the processed PPco as compared to that of as-received PPco confirmed our assumption. During the TGA testing, both 380 °C and 420 °C peaks appeared for the TDTG-PPco. This reflects the different processing history of PPco because some PPco were added before and some PPco were added after the masterbatch process. Samples with lower TDTG-PPco are analyzed here because they are more likely to be degraded in the next printing process. The addition of SDCNF increased the residual mass content of PPco at 450 °C. This implies that SDCNF slightly hinders the thermal degradation of PPco at higher temperature [19]. The purpose of performing TGA was to predict whether or not the thermal degradation occurred during the AM process. During AM, filaments flow continuously through the liquefier for a short residence time. Moreover, much smaller shear rate was reported during extrusion-based AM as opposed to injection molding [23]. Additionally, the polymer was printed at 200 °C. Based on this information, no severe thermal degradation was expected during AM printing.

TGA (a) and DTG (b) graphs of PPco, SDCNF and SDCNF-PPco composites
Differential scanning calorimetry (DSC)
DSC curves of all pellets displayed similar trend during the tests, only the curves of neat PPco pellets are shown in Fig. 3 for illustration. The related thermal properties are listed in Table 3. PPco and SDCNF-PPco composites all have a Tm around 160 °C which corresponds to the melting of α crystals [3]. SDCNF does not appear to change the Tm of PPco, which was also reported in previous research [14,15,16,17]. The addition of SDCNF gradually increases the Tc of PPco, indicating SDCNF, under current testing conditions, acts as a nucleating agent for PPco. In a previous study, CNF was reported to be a nucleating agent, which increased the Tc of PP [15]. The RXc of PPco with SDCNF is larger than pure PPco, which again confirms the nucleating ability of SDCNF for PPco. Generally, the addition of MAPP into either PPco or SDCNF-PPco composites promotes the RXc of PPco. First, MAPP alone was reported to be a nucleating agent for PP [24, 25]. Second, MAPP facilitates SDCNF to distribute better within PP, which increases the nucleation density. Based on above information, a printing temperature of 200 °C should be sufficient for melting the PPco and its composites. A bed temperature of 120 °C will improve the RXc of PPco as it constantly anneals the specimens during printing. An increase in Xc of PLLA was reported using a bed temperature of 160 °C [26].

Melting (a) and crystallization (b) processes of PPco pellets measured by DSC
After samples are processed by injection molding or additive manufacturing, their melting behavior differed significantly. The melting curves of injection molded PPco (PPcoIM) and AM printed PPco (PPcoAM) are shown in Fig. 4. In Table 4, only PPcoIM was injection molded, all others were additive manufactured. For PPcoIM, only one melting peak was observed at around 160 °C which is the melting temperature of α crystal of PP [27]. For PPcoAM, they all show three peaks at about 145 °C, 150 °C and 160 °C, corresponding to the β, β′ and α crystals of PP [3, 27]. The formation of β crystal can be promoted by a controlled crystallization temperature range (100–120 °C), β-nucleating agent, directional crystallization in a temperature gradient field and shear-induced crystallization [28]. Based on our previous study of 3D printed PP, a controlled crystallization temperature is most likely the major reason for the formation of β crystal in this study as the printing bed temperature was kept at 120 °C [3]. β′ crystal is formed when less stable β recrystallizes to more stable α crystal. β′ crystal is a transitional phase that is caused by the heating used in the DSC test [29], thus should not be used for calculating crystallinity. To calculate the RXc for PPcoAM, Eq. (1) cannot be used. Instead, the Xα and Xβ ought to be calculated separately because α and β crystals have different heats of fusion [30]. The contents of β and α crystal affect the mechanical properties of PP [31]. Because the mechanical properties are not the interest of this study, as well as the complexity of calculations, Xc, Xα and Xβ were not computed for PPcoAM samples.

Melting curve of injection molded (IM) and additive manufactured (AM) PPco
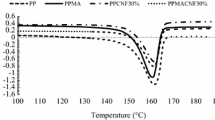
Heat deflection temperature (HDT)
HDTs of injection molded PPco and printed PPco composites are displayed in Table 5 where only PPcoIM was injection molded, and all others were additive manufactured. PPcoAM samples have a significantly higher HDT than PPcoIM. Despite the formation of voids in the PPcoAM samples, they displayed the existence of β crystals (Fig. 4) which were reported to improve the HDT of PP [32]. Among the AM printed groups, PPcoMA has a higher HDT than PPco. HDT of a solid polymer is related to the Tm and Xc of the material [33]. Because MAPP can increase the RXc of PPco (Table 3), the addition of MAPP increases the HDT of PPco. Adding SDCNF increases the HDT of printed PPco. There are two possible explanations for the observed behavior: (1) It was reported that natural fibers in PP restricted the mobility of PP chains in the amorphous area, leading to a higher HDT [34, 35]. (2) As seen in Fig. 5, the addition of SDCNF increased the number of voids in printed parts. It was reported that the addition of carbon fibers also increased the void portion in 3D-printed thermoplastics [36]. The sources of void formation during 3D printing include incomplete diffusion among laid-down strands (Fig. 5b), off-gassing and fiber pullout [36]. The part voids acted as insulator that delayed heat transfer, thus increasing the HDT. An increase in HDT of foamed willow-fiber-filled PLA was reported compared to unfoamed injection molded PLA [37]. However, these voids will degrade the mechanical properties of printed parts, thus should be avoided to make better quality parts.

SEM graphs of injection molded PPco (a), printed PPco (b), printed PPcoSDCNF7.5 (c) and printed PPcoSDCNF15 (d). White dots in the micrographs are SDCNF
Conclusions
Thermal analysis was performed on PPco and SDCNF-PPco composite pellets and specimens.
-
1.
TGA on PPco pellets showed that the addition of SDCNF lowered the onset temperature and increased the residual mass content (at 450 °C) of PPco pellets without significantly changing the peak degradation temperatures of SDCNF and PPco.
-
2.
SDCNF and MAPP increased the peak crystallization temperature and relative crystallinity of PPco pellets without changing its melting temperature. This indicated that SDCNF and MAPP were nucleating agents for PPco under the test conditions.
-
3.
3D-Printed PPco and SDCNF-PPco composites exhibited three melting peaks that belong to β, β′ and α crystals while injection molded PPco only showed a melting peak of α crystal. A bed temperature of 120 °C was the major reason for the formation of β crystal in printed parts.
-
4.
Heat deflection tests showed that printed PPco had higher HDT than injection molded PPco because 3D-printed PPco has β crystals. 3D-Printed SDCNF-PPco composites had higher HDT than 3D-printed PPco because SDCNF increased the number of voids and restricted the mobility of PP chain in amorphous areas.
References
Wendel B, Rietzel D, Kühnlein F, Feulner R, Hülder G, Schmachtenberg E. Additive processing of polymers. Macromol Mater Eng. 2008;293:799–809.
Wang L, Gardner DJ, Bousfield DW. Cellulose nanofibril-reinforced polypropylene composites for material extrusion: rheological properties. Polym Eng Sci. 2017. https://doi.org/10.1002/pen.24615.
Wang L, Gardner DJ. Effect of fused layer modeling (FLM) processing parameters on impact strength of cellular polypropylene. Polymer. 2017;24:74–80.
Nandi S, Ghosh AK. Crystallization kinetics of impact modified polypropylene. J Polym Res. 2007;14:387–96.
Moon RJ, Martini A, Nairn J, Simonsen J, Youngblood J. Cellulose nanomaterials review: structure, properties and nanocomposites. Chem Soc Rev. 2011;40:3941–94.
Wang L, Sanders JE, Gardner DJ, Han Y. In-situ modification of cellulose nanofibrils by organosilanes during spray drying. Ind Crops Prod. 2016;93:129–35.
Gardner DJ, Han Y, Wang L. Wood–plastic composite technology. Curr For Rep. 2015;1(3):139–50. https://doi.org/10.1007/s40725-015-0016-6.
Wang L, Roach AW, Gardner DJ, Han Y. Mechanisms contributing to mechanical property changes in composites of polypropylene reinforced with spray-dried cellulose nanofibrils. Cellulose. 2018;25(1):439–48. https://doi.org/10.1007/s10570-017-1556-7.
Sun L, Wu Q, Xie Y, Song K, Lee S, Wang Q. Thermal decomposition of fire-retarded wood flour/polypropylene composites. J Therm Anal Calorim. 2016;123(1):309–18.
Tajvidi M, Takemura A. Thermal degradation of natural fiber-reinforced polypropylene composites. J Thermoplast Compos Mater. 2010;23:281–98.
Zhao P, Guo C, Li L. Flame retardancy and thermal degradation properties of polypropylene/wood flour composite modified with aluminum hypophosphite/melamine cyanurate. J Therm Anal Calorim. 2018. https://doi.org/10.1007/s10973-018-7544-9.
Tajvidi M, Sharma AM, Simon LC. Thermal transitions and temperature dependent mechanical behavior of wheat straw/talc isotactic/impact modified polypropylene composites. J Reinf Plast Compos. 2013;32:1430–43.
Guo B, Zhang T, Zhang W, Dou Y. Influence of surface flame-retardant layer containing ammonium polyphosphate and expandable graphite on the performance of jute/polypropylene composites. J Therm Anal Calorim. 2018. https://doi.org/10.1007/s10973-018-7406-5.
Ljungberg N, Cavaillé JY, Heux L. Nanocomposites of isotactic polypropylene reinforced with rod-like cellulose whiskers. Polymer. 2006;47:6285–92.
Panaitescu DM, Donescu D, Bercu C, Vuluga DM, Iorga M, Ghiurea M. Polymer composites with cellulose microfibrils. Polym Eng Sci. 2007;47:1228–34.
Nagalakshmaiah M, El Kissi N, Dufresne A. Ionic compatibilization of cellulose nanocrystals with quaternary ammonium salt and their melt extrusion with polypropylene. ACS Appl Mater Interfaces. 2016;8:8755–64.
Bahar E, Ucar N, Onen A, Wang Y, Oksüz M, Ayaz O, Ucar M, Demir A. Thermal and mechanical properties of polypropylene nanocomposite materials reinforced with cellulose nano whiskers. J Appl Polym Sci. 2012;125:2882–9.
Hassan ML, Mathew AP, Hassan EA, Fadel SM, Oksman K. Improving cellulose/polypropylene nanocomposites properties with chemical modified bagasse nanofibers and maleated polypropylene. J Reinf Plast Compos. 2014;33:26–36.
Yang HS, Kiziltas A, Gardner DJ. Thermal analysis and crystallinity study of cellulose nanofibril-filled polypropylene composites. J Therm Anal Calorim. 2013;113:673–82.
Khoshkava V, Kamal MR. Effect of cellulose nanocrystals (CNC) particle morphology on dispersion and rheological and mechanical properties of polypropylene/CNC nanocomposites. ACS Appl Mater Interfaces. 2014;6:8146–57.
González-González VA, Neira-Velázquez G, Angulo-Sánchez JL. Polypropylene chain scissions and molecular weight changes in multiple extrusion. Polym Degrad Stab. 1998;60:33–42.
Hermanová S, Tocháček J, Jančář J, Kalfus J. Effect of multiple extrusion on molecular structure of polypropylene impact copolymer. Polym Degrad Stab. 2009;94:1722–7.
Turner BN, Strong R, Gold SA. A review of melt extrusion additive manufacturing processes: I. Process design and modeling. Rapid Prototyp J. 2014;20:192–204.
Seo Y, Kim J, Kim KU, Kim YC. Study of the crystallization behaviors of polypropylene and maleic anhydride grafted polypropylene. Polymer. 2000;41:2639–46.
Wang L, Gramlich WM, Gardner DJ, Han Y, Tajvidi M. Spray-dried cellulose nanofibril-reinforced polypropylene composites for extrusion-based additive manufacturing: nonisothermal crystallization kinetics and thermal expansion. J Compos Sci. 2018;2(1):7. https://doi.org/10.3390/jcs2010007.
Wang L, Gramlich WM, Gardner DJ. Improving the impact strength of poly (lactic acid)(PLA) in fused layer modeling (FLM). Polymer. 2017;114:242–8.
Tordjeman P, Robert C, Marin G, Gerard P. The effect of α, β crystalline structure on the mechanical properties of polypropylene. Eur Phys J E Soft Matter. 2001;4:459–65.
Papageorgiou DG, Chrissafis K, Bikiaris DN. β-nucleated polypropylene: processing, properties and nanocomposites. Polym Rev. 2015;55:596–629.
Fujiwara Y. Das doppelschmelzverhalten derb-Phase des isotaktischen polypropylenes. Colloid Polym Sci. 1975;253:273–82.
Li JX, Cheung WL, Jia D. A study on the heat of fusion of β-polypropylene. Polymer. 1999;40:1219–11222.
Wang L, Sanders JE, Gardner DJ, Han Y. Effect of fused deposition modeling process parameters on the mechanical properties of a filled polypropylene. Prog Addit Manuf. 2018. https://doi.org/10.1007/s40964-018-0053-3.
Varga J. β-modification of isotactic polypropylene: preparation, structure, processing, properties, and application. J Macromol Sci B. 2002;41(4–6):1121–71.
Landel RF, Nielsen LE. Mechanical properties of polymers and composites. Amsterdam: CRC Press; 1993.
Chattopadhyay SK, Khandal RK, Uppaluri R, Ghoshal AK. Mechanical, thermal, and morphological properties of maleic anhydride-g-polypropylene compatibilized and chemically modified banana fiber-reinforced polypropylene composites. J Appl Polym Sci. 2010;117:1731–40.
Chattopadhyay SK, Khandal RK, Uppaluri R, Ghoshal AK. Bamboo fiber reinforced polypropylene composites and their mechanical, thermal, and morphological properties. J Appl Polym Sci. 2011;119:1619–26.
Ning F, Cong W, Qiu J, Wei J, Wang S. Additive manufacturing of carbon fiber reinforced thermoplastic composites using fused deposition modeling. Compos Part B Eng. 2015;80:369–78.
Zafar MT, Zarrinbakhsh N, Mohanty AK, Misra M, Maiti SN, Ghosh AK. Biocomposites based on poly (lactic acid)/willow-fiber and their injection moulded microcellular foams. Expr Polym Lett. 2016;10:176–86.
Acknowledgements
Funding was provided in part by the National Science Foundation REU Project No. 1461116, the Maine Agricultural and Forest Experiment Station (MAFES) project ME0-M-8-00527-13 and the USDA ARS Forest Products Research Agreement 58-0202-4-003. The authors thank J. Elliott Sanders at the University of Maine for preparing the pellets and composites for analysis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Wang, L., Palmer, J., Tajvidi, M. et al. Thermal properties of spray-dried cellulose nanofibril-reinforced polypropylene composites from extrusion-based additive manufacturing. J Therm Anal Calorim 136, 1069–1077 (2019). https://doi.org/10.1007/s10973-018-7759-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-018-7759-9