
Abstract
Refined structure of the ferritic phase induced by mechanical milling (under reducing atmosphere) and its thermal stability are required in various applications of nanostructured ferritic alloys. The impurification with nitrogen and oxygen uptaken from the air is very probable during ball-milling, especially at the long-time high-energy milling conditions. As a rule, these interstitial impurities in as-milled powders are in quantities under the sensibility limit of conventional measurement techniques, such as XRD and SEM/EDS. To evidence the tendency for microstructure modification by impurities introduced during milling, the Fe–14Cr–3W–0.4Ti–0.25Y2O3 (Fe14Cr) ferritic steel powders (re)loaded in air and milled up to 170 h with interruption of the milling process, and heated up to 1373 K were investigated by thermal analysis in correlation with X-ray diffraction and scanning electron microscopy. XRD failed to detect the impurities in powders milled up to 38 h in air although a consistent mass loss related to the degassing of N2 was registered in thermogravimetric, TG, curves. (Fe,Cr)4N, fcc-γ, (Fe,Cr)2O3 impurity phases in powders milled over 38 h in air and (Fe,Cr)2O4 formed upon heating were readily detected by XRD. The analysis of these results allowed to better understand the impurification process and to generalise it for any as-milled Fe–Cr-based alloy powder processed in any milling conditions irrespective of the milling atmosphere, duration and thus, of amounts of contaminants. The quality of three powders milled for 170 h in three different conditions was compared: in air, under an argon atmosphere with interruptions of the milling process and under an argon atmosphere without interruption of the milling process. The contamination of powder milled for 170 h under an argon atmosphere without interruption of the milling process is insignificant (corresponding to less than 0.5 mass% mass loss in TG) as compared to powders obtained in the other two milling conditions. New approaches for minimising the contamination from air are suggested.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The microstructure, phases and thermal stability are important conditions for the broad range of applications of ferritic stainless steels. This also relates to the oxide dispersion strengthened ferritic steels (ODSFS) which are very promising for applications in fusion and fission power reactors [1]. Particularly, reduced activation Fe–Cr–W–Ti–Y2O3 ODSFSs are under intense development as materials with superior high-temperature mechanical properties relevant for breeding blanket structures of the DEMO (Demonstration Fusion Power Reactor) [1, 2]. Refined structure induced by mechanical milling [3] is required in this and other applications. During milling, the process of the impurification of powders with residual nitrogen and oxygen pre-existent on the container walls/balls and/or uptaken from the air (when the mill operates under atmospheric conditions) is nearly inherent [3, 4]. Moreover, the initial powders themselves contain impurities especially on the powder particle surface [5,6,7], particularly in the form of oxide passivation layer [5, 6]. During milling in the presence of the impurities, they can be easily absorbed and/or pinned at the created defects. Particularly, they can enter interstitially [8,9,10] or even can generate secondary impurity phases especially at long-time high-energy milling conditions [11]. The impurities are able to influence the microstructure of steels even when they are in quantities under the detection limit of conventional measurement techniques, such as XRD and SEM/EDS. The as-milled metal alloy powder, an extremely complex metastable system, is far from being well understood. As about the influence of contamination during milling (in fact uncontrolled factor) on the microstructure properties of Fe–Cr-based alloys, very few investigations were carried out [11,12,13].
In our previous work [11], the process of the contamination from the air during milling with interruption of the milling process of Fe–14Cr–3W–0.4Ti–0.25Y2O3 ferritic steel powders (re)loaded under an Ar atmosphere was investigated. The traces of reflections corresponding to a new phase with an fcc-CrN structure appeared in XRD of the as-milled powders for long-time milling, 113–170 h. However, for shorter milling time no impurity phases were observed by XRD and EDX. Instead, the methodology of the thermal analysis was successfully applied for the registration of the impurity phases generated by contamination of these powders. In particular, upon DTA heating an exothermic feature at 898–926 K was associated with the precipitation or coarsening of nitrides and an endothermic feature at 1198–1523 K accompanied by a mass loss was ascribed to the decomposition of nitrides with degassing of N2. In addition, we demonstrated that the contamination level depends on the degree of alloying at the moment of the interruption of the milling process. Within this work, the evolution of the microstructure of the Fe–14Cr–3W–0.4Ti–0.25Y2O3 ferritic phase and of the impurity phases upon milling (by (re)loading) in air and upon the heating of the as-milled powders was investigated. For shorter milling time, below 38 h, were XRD fails to detect the impurity phases the mass loss and, exothermic and endothermic features registered in TG/DTA evidence the presence of contaminants from the air. This result together with the large amounts of impurity phases in powders milled over 55 h in air readily detected by XRD, facilitated the understanding and the interpretation of the impurification process irrespective of the milling conditions. The quality of powder milled for 170 h in air and, of powders milled for 170 h under an Ar atmosphere with and without the interruption of milling process was compared. Finely, a new approach for minimising the contamination from the air was suggested. The results of this work can be generalised to any Fe–Cr-based ball-milled ferrite powder.
Experimental
The details of sample preparation are given in [11]. Briefly, elemental powders of iron, chromium, tungsten, titanium, and nano-yttrium oxide were weighted in the ratios of Fe/Cr/W/Ti/Y2O3–82.35:14:0.4:0.25 in mass% and thoroughly mixed. The mixture of elemental powders was mechanically alloyed in a Retch planetary ball mill, the ratio of the mass of the balls to the powder (BPR) was 10:1 and the milling speed was 500 to 550 rpm.
The description of the milling conditions of the powders milled in air is given in Table 1. The batch C powders C0.17, C0.5, C1, C2, C3, C4, C6, C12, C24, C38, C55, C72, C113 and C170 were obtained by loading the mixture of initial elemental powder, C0, into the vial in air and milling for total durations of 0.17, 0.5, 1, 2, 3, 4, 6, 12, 24, 38, 55, 72, 113 and 170 h, respectively. At each interruption of the milling process (Table 1), the vial was opened in air, a quantity of approximately 250 mg of the powder was taken out for further characterisations after that the vial was resealed in air. The powders of batch A (A0.17, A0.5, A1, A2, A3, A4, A6, A12, A24, A38, A55, A70, A113 and A170) were obtained similarly to the powders of batch C but with operation under the Ar atmosphere as detailed in the Ref. [11]. In particular, the powder A170 was obtained (as described in [11] and in Table 1) by loading the mixture of initial elemental powders into the vial under an argon atmosphere and milling for a total duration of 170 h with 13 interruptions the milling process: the vial was opened and resealed under an argon atmosphere. The batch B powders B6, B12, B38, B70 [11] and B170 were obtained by loading the mixture of initial elemental powders into the vial under an argon atmosphere and milling them for 6, 12, 38, 70 and 170 h, respectively, without interrupting the milling process.
In order to investigate the phase evolution upon heating, the as-milled powders were heat treated in a quartz tube furnace for up to 5 min at different temperatures (593–1423 K) in an atmosphere of 95% Ar and 5% H2 which was then quenched to room temperature. The heating rate was 10 K min−1.
Thermal analysis was performed using a modular simultaneous thermal analyser SETARAM Setsys Evolution 18 apparatus coupled with a quadrupole mass spectrometer, MS, (Pfeiffer Vacuum). About 150 mg (weighted with a precision of ± 10−2 mg) of as-milled powders was loaded into an open cylindrical alumina crucible. The experiments were conducted in Ar of 5N (99.999%) purity at a flow rate of 16 mL min. The temperature scan was performed between 298 and 1373 K/1623 K at a heating and cooling rate of 10 K min−1 (without isothermal holding) with a temperature precision of better than 0.01 K. From the DTA heating curve, the peak of the Curie temperature and α→γ transition temperature was also determined.
Qualitative and quantitative phase analysis and the microstrain and crystallite size (coherence length) estimation were performed on a Bruker D8 Advance diffractometer in Bragg–Brentano geometry using a copper target X-ray tub. The XRD data were processed by Rietveld whole powder pattern fitting for the as-milled and heat-treated powders using the Bruker-TOPAS software and the fundamental parameter approach (FPA). The combined effect of size and microstrain upon the line profile was taken into account only for the majority phase (Fe–Cr), which showed well-defined diffraction line profiles. The microstrain was neglected for the minor phases (e.g. nitrides and oxides), leading to the possible underestimation of sizes.
Results and discussions
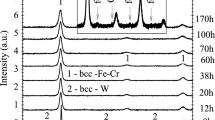
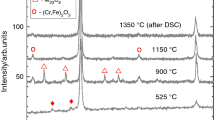
The X-ray diffraction patterns for batch C powders milled in air after different milling times, t, are shown in Fig. 1a, b. Up to 24 h milling, they reveal an exclusive phase of bcc-α structure, and no secondary impurity phases were detected. The Cr diffraction lines disappeared, whereas the intensity of W reflections diminished drastically at 12 h of milling, indicating that the alloying elements had diffused into the Fe matrix. At 38 h of milling, a trace of oxides with (Fe,Cr)2O3 structure appears, and its quantity increases with increasing milling time. The lattice parameters were determined from the bcc-Fe–Cr diffraction peaks positions, which, as shown in Fig. 1, broaden and shift to lower angles with the advancing in milling process. The enlarged view of the diffraction peak at 2Θ ≈ 44.65 (Fig. 1b) shows that the broadening is caused in particular by formation of a phase of fcc-γ structure detected at 55 h and increasing with increasing milling time up to 170 h. The evolution of the powders’ grain size and microstrain as a function of the milling time is shown in Fig. 2a, b, respectively. The lattice constant versus milling time is shown in Fig. 2c. As expected, mechanical alloying resulted in the increase in the lattice parameter and of the microstrain, and decrease in the crystallite size. The SEM micrographs of the as-milled powders are shown in Fig. 3. Figure 4 shows the TG/DTA curves for batch C powders milled in air for different durations. The dependence of TG mass (nitrogen) loss on the milling time is shown in Fig. 5. The X-ray diffraction patterns for the C170 powder of batch C after heating at different temperatures are shown in Fig. 6. In Table 2, there are listed the microstructure parameters of the ferritic phase and of the secondary phases at different milling times for the C170 product strongly contaminated with nitrogen and oxygen. Figure 7 shows the evolution of the quantity of the impurity phases in C170 powder (milled for 170 h in air with interruptions) with increasing heating temperature. The XRD and TG/DTA curves for A170 and B170 powders milled in an argon atmosphere without and with interruption of milling process are shown in Figs. 8 and 9, respectively, together with C170 powder milled in air. Figure 9b also presents the mass spectra for m/z 28 of A170 and B170 (dashed line).

XRD patterns describing the evolution of structure and phases composition with increasing milling time up to 170 h of powders milled in air with interruption. The most intense Bragg 110 peak of bcc-Fe–Cr was normalised to unity for all of the samples. The arrows highlight the presence of a shoulder corresponding to Cr rich zones. Inset: magnified view for 110 peak at 2Θ ≈ 44.65

The a crystallite size (coherent length), b microstrain and c the lattice constant versus milling time of powders milled in air with interruption

SEM images describing the evolution of powder morphology with increasing milling time: a C0 (mixture of elemental powders), b C0.5, c C3, d C6, e C72 and f C113 powders milled in air with interruption for the total duration of 0, 0.5, 3, 6, 72 and 113 h, respectively

a DTA and b TG curves measured on Fe–14Cr–3W–0.4Ti–0.25Y2O3 powders milled in air with interruption. The heating rate was 10 °C min−1

Evolution of the mass loss with milling time after DTA heating to 1100 °C of powders milled in air with interruption. At 55 h the rate of nitrogen uptake from the air changes from fast to slow regime. The fit of this two regimes with two linear functions gives the slops corresponding to the rate of nitrogen uptake from the air upon milling: d(N)/dt = 0.042 mass% h−1 and d(N)/dt = 0.0132 mass% h−1

XRD patterns of C170 powder (milled in air with interruption for the total duration of 170 h) as-milled and furnace heated to 320, 485, 675, 750, 950, 1040 and 1150 °C in Ar 5% H2 atmosphere. The most intense Bragg 110 peak of bcc-Fe–Cr was normalised to unity for all of the samples

The evolution of the quantity of impurity phases fcc-γ, (Fe,Cr)4N, (Cr, Fe)2O, and (Fe,Cr)2O4 in C170 powder (milled in air for 170 h with interruptions) with increasing heating temperature

XRD patterns of A170 milled under an Ar atmosphere with interruption, B170 milled under an Ar atmosphere without interruption and C170 milled in air with interruption. The most intense Bragg 110 peak of bcc-Fe–Cr was normalised to unity for all of the samples

a DTA and b TG curves measured on Fe–14Cr–3W–0.4Ti–0.25Y2O3 powders: A170 milled under an Ar atmosphere with interruption, B170 milled under an Ar atmosphere without interruption and C170 milled in air with interruption. In b the mass spectra for m/z 28 of A170 and B170 are also shown (dashed line). The heating rate was 10 °C min−1
Microstructure modification by milling in air with the increasing of milling time
By milling up to 6 h in air the rapid increase in lattice constant, l (from 2.8662 Å for C0 to 2.8725 Å for C6), and microstrain, <ε2>1/2 (from 0.023% for C0 to 0.278% for C6), and, rapid decrease in crystallite size, d (from 103 nm for C0 to 16.5 nm for C6), is observed (Fig. 2). Such modification of microstructure parameters, attributed to the creation and accumulation of dislocations, stacking faults, high-angle GBs, interfaces and other defects and, to the mixing between the alloying elements [12, 13], was induced by severe plastic deformation and high fracturing rate of powder particles at this stage of milling. Random distribution of stress fields created by these defects resulted in microstresses and thus, in increase in microstrain. The decrease in grain size is primarily associated with the increase in density of grain boundaries (GBs). The increase in the degree of Fe–Cr alloying correlates with the observed strong lattice constant increase for the powders milled to 6 h. The described microstructure modification is in agreement with the observed modification of the powder morphology as shown in Fig. 3. Similarly to the milling in an Ar atmosphere described in details in [11], in the first 1 h of milling in air the particles fracture and flatten due to the process of severe plastic deformation at the initial stage of milling (Fig. 3b). Upon increasing the milling time to 6 h (Fig. 3c), the aspect ratio of the particles decreases. However, the presence of a large number of the particles with very small size (under 1 μm) suggests a still high fracture rate at this stage of milling in air.
Between 6 and 55 h of milling, the microstructure parameters change more slowly (Fig. 2). The observed relative saturation in the grain size decreases, and the saturation in the increase in microstrain and lattice constant can be explained by the fact that the density of defects (e.g. dislocations, GBs [14]) approached saturation [15]. The saturation is associated with achieving a balance between the rate of welding and the rate of fracturing the powder particles [11]. Nonetheless, a still increasing microstrain (from 0.278% for C6 to 0.435% for C55) and lattice constant (from 2.8724 Å for C6 to 2.8738 Å for C55) and decreasing grain size (from 16.5 nm for C6 to 6.9 nm for C55) when milling between 6 and 55 h can be explained by the additional defects generated by nitrogen and oxygen uptaken from the air during milling and thus by the presence of solute nitrogen/oxygen in the ferritic phase. Traces of contaminants, the reflections of (Cr, Fe)2O3 oxide structure, appeared in XRD at 38 h milling (Fig. 1). Even though the contamination of the powders milled up to 38 h was not detected by XRD (Fig. 1), the contamination was evident from thermal analysis (Fig. 4). The increase in the quantity of TG mass loss with increasing milling time caused by nitrogen degassing is very evident from Fig. 4b.
As shown in Fig. 2, specific to the powders milled over 55 h is a high rate of microstrain increase (from 0.435% for C55 to 1.25% for C170) and lattice constant increase (from 2.8738 Å for C55 to 2.9246 Å for C170). The grain size of the powders milled over 55 h was further reduced and attained very fine size, 4.5 nm, for C170. Such refinement denotes a highly disordered, up to the limit of amorphous, structure which can explain the excessively high values of the microstrain, 1.25%, for C113 and C170. The pronounced modification of the microstructure parameters of the ferritic phase when milling for long time in air (Figs. 1b, 2) is due mainly to the supersaturation of the ferritic phase with nitrogen entered interstitially into the lattice. An explanation for the structure refinement (the ultrafine grain size) of the ferrite is that interstitial impurities trapped by GBs and dislocations [10] during milling pinned them, impeding dynamic recrystallisation and dislocation mobility. Similar refinement effects were reported on Fe [8, 9] and Fe-based powders milled together with iron nitrides [4] and/or under a nitrogen atmosphere [16]. The substantial modification of microstructure parameters of the ferritic phase when milling above 55 h is supported by drastic modification of the phase composition of the steel as clearly shown in Fig. 1a. Phases of fcc-γ structure and Fe4N structure have precipitated at the milling stage in C55, C72 and C113 and, C170, respectively (Fig. 1). The contamination and supersaturation with nitrogen was estimated from the TG curves (as described in more details in next section) to be about 3.8 mass% for C170 (Fig. 5), well above the equilibrium solubility limit of nitrogen in iron at room temperature (~ 0.1 mass% [17]). The high affinity of Cr alloying element to the nitrogen improved its solubility within the steel matrix promoting as well the precipitation of the excess nitrogen atoms as (Fe,Cr)4N and fcc-γ phase. The α→γ transformation was favoured during high-energy milling in the presence of nitrogen because [4, 16]: nitrogen is a strong austenite stabiliser element (nitrogen atoms create less distortion in austenite than in ferrite); the process of refinement by nitrogen favours transition to austenite because the grained fcc structure has smaller interfacial energy compared to bcc ferritic structure. Thus, the pronounced decrease in the volume fraction of bcc-α phase during milling in air (70.6 mass% for C170) is caused by the precipitation/segregation of a large amount of secondary phases—(Fe,Cr)4N (6.3 mass% for C170), fcc-γ (10.9 mass% for C170) and (Fe,Cr)2O3 (12.3 mass% for C170) (Fig. 1, Table 2). It is worth to mention that no decrease in lattice constant was observed, as it was expected after the extraction of Cr form the ferrite lattice by precipitation/segregation of these secondary phases. On the contrary, the pronounced increase in lattice constant of bcc-α for the powders milled for 55–170 h (Fig. 2c) is consistent with the (metastable) ferritic phase supersaturated with nitrogen eventually coexisting with the above-mentioned secondary phases.
It is worth noting that in contrast to powders milled over 6 h under an Ar atmosphere with interruption of milling process [11] showing the increase in the powder particle size, the particle size does not increase by milling in air over 6 h (Fig. 3). This can be explained by the fact that the fracturing rate remains permanently high because the particles harden due to the progressive contamination with large amounts of nitrogen (Figs. 1, 5) [and oxygen (Fig. 1)] as the milling time increases [15]. In addition, the contaminants from the air act as a process control agent during milling. The crystallite size of batch C powders milled in air for long time with interruptions (e.g. 16.5 nm at the steady state and 4.1 nm after 170 h milling.) is smaller as compared to batch A powders milled under an Ar atmosphere with interruptions (e.g. 18.1 nm at the steady state, 15.0 nm after 170 h milling).
Thermal analysis—Heat treatment
As seen from DTA curves for batch C powders milled in air (Fig. 4a) besides the endothermic peak 2 at 1025–1045 K corresponding to magnetic transition (Curie temperature, Tc) and endothermic peak 3 at 1184–1223 K corresponding to α→γ transition another endothermic peak 1′ at 897–915 K was registered for the powders milled over 38 h (38–170 h). This endothermic feature is accompanied by a mass loss in TG in ~ 873–1093 K temperature range. Another step of mass loss in TG is observed above 1093 K for 3–72 h milled products, which accompanies an endothermic feature 4 observed for 12–55 h milled powders. Further discussions of the results of thermal analysis in this and in the next subsection will be made in correlation with the results of furnace heating of most contaminated powder C170 obtained by milling in air for 170 h—the high amount of the impurity phases readily detected by XRD in this as-milled and heated product facilitates the understanding of the impurification process in other samples irrespective of milling atmosphere, duration and thus, of the amount of contaminants.
If we follow the TG curve of C170 powder (Fig. 4b) in correlation with the evolution of the quantity of impurity phases during furnace heating (estimated from XRD patterns and illustrated in Fig. 7), then it becomes clear that the observed TG mass losses when heating up to ~ 1373 K were caused by N2 degassing when the secondary phases containing nitrogen ((Fe,Cr)4N, fcc-γ) decomposed. As shown in Fig. 5, the evolution of the quantity of TG mass (nitrogen, N) loss with increasing milling time of the batch C powders occurred in two regimes, similarly to the modification of microstructure parameters (Figs. 2, 4): (1) high rate regime (0.042 mass% h−1)—up to 2.42 mass% total mass loss for powders milled up to 55 h and low rate regime (0.013 mass% h−1)—2.42–3.9 mass% mass loss for powders milled over 55 h.
At the early stage of heating up to 500 °C, the powders underwent recovery, recrystallisation, and initiation of grain growth processes [14, 15, 18]. The decrease in microstrain and lattice parameter (Table 2) when heating in this region is attributable to the decreased number of defects (e.g. dislocations). At about 773 K, the microstain and lattice parameter relaxed completely and they does not change anymore with farther increasing temperature. The pronounced grain growth occurs above 773 K [from 8.8 nm at 758 K to 196.3 nm at 1423 K for C170 (Table 2)]. The processes of the decomposition, dissolution or reduction of the metastable secondary phases formed during milling ((Fe,Cr)4N, fcc-γ and (Fe,Cr)2O3) as well as the nucleation, precipitation or reoxidation [11, 19] of new secondary phases, e.g. (Fe,Cr)2O4, (Figs. 6, 7, Table 2) accompany the processes of recovery and recrystallisation of ferritic phase and ferrite grain growth. fcc-γ decomposed during relaxation when heating up to about 958 K. The metastable (Fe,Cr)4N formed during milling initially stabilised up to about 773 K heating after that it decomposed rapidly up to about 958 K (Figs. 6, 7). This comportment of the secondary phases correlates with the above-mentioned endothermic feature 1′ in DTA, and the associated maximum in energy consumption for C170 is registered at 915 K (or in the 853–973 K temperature range for C55–C170). The decomposition of fcc-γ and (Fe,Cr)4N is accompanied by the nitrogen degassing associated with the above-mentioned pronounced step in the TG curves below 1093 K for C55–C170 (Fig. 4b). These features are followed by an exothermic peak 1″ registered at 953–998 K for C55–C170 which might be ascribed to the enhancement of Fe–Cr solid solution by Cr and Fe realised after the decomposition of above-mentioned secondary phases.
As for (Fe,Cr)2O3 its quantity decreases considerably by heating above 1023 K. The known high affinity of Cr to oxygen can drive the oxidation process even when treated under an inert and reducing atmosphere [5,6,7]. In this context, the reduction of (Fe,Cr)2O3 segregated during milling denotes its high instability in the given conditions of treatment and phases compositions. Upon heating, the unstable (Fe,Cr)2O3 is reduced [20,21,22,23] under inert atmosphere to the more stable (Fe,Cr)2O4. (Fe,Cr)2O4 appears at 593 K, its quantity increases significantly above 1223 K in good agreement with the reduction of (Fe,Cr)2O3. Upon heating over 1223 K, the (Fe,Cr)2O3 is reduced completely to (Fe,Cr)2O4.
The contamination under different milling conditions
As mentioned hereinbefore, the mass loss in TG occurs in two temperature regions, up to 1093 K in the ferrite phase field and above 1093 K in the austenite phase field (Fig. 4b). The TG mass loss below 1093 K was registered for 38–170 h products milled in air (batch C) and relates to nitrogen degassing after the decomposition of metastable fcc-γ and (Fe,Cr)4N phases formed during milling. These impurity phases are well visible in XRD for 55–170 h milled powders (Fig. 1). The TG mass loss above 1093 K was registered for 3–72 h milled powders in air (batch C). Similarly to less contaminated batch A powders (milled under and Ar atmosphere with the interruption of milling process, described in Ref. [11]) TG mass loss above 1093 K relates to N2 degassing after the decomposition of residual phases containing nitrogen which are stable in the ferrite phase field, namely the phases that have precipitated in small quantities during milling or have precipitated/stabilised upon heating (this will be more clear from the discussed hereinafter).
The discussed in the previous paragraph can be generalised to all powders of batches A, B and C as shown hereinafter. Figures 8 and 9 present the results of XRD and TG/DTA, respectively, for three representative powders milled for 170 h in three different conditions—in air (C170), under an argon atmosphere with interruption of the milling process (A170) and under an argon atmosphere without interruption of the milling process (B170). The powders of batches A, B and C can be divided into three categories represented by A170, B170 and C170. Any Fe14Cr as-milled powders can be assigned to one of these three categories depending on TG/DTA behaviour as well as on amounts of impurity phases and their behaviour during heating. C170-category comprises the highly contaminated powders C72–C170. A170-category comprises moderately contaminated powders A70–A170 [11] and C38–C55. B170-category comprises weakly contaminated powders A0.5–A55 [11], B6–B70 [11], B170 and C0.5–C24. As already discussed, the mass loss of powders of C170-category occurs in the ferritic phase field below 820 °C due to nitrogen degassing after the decomposition of metastable impurity phases formed during milling. For weakly contaminated products of B170-category and for moderately contaminated products of A170-category a mass loss is registered above 1093 K in austenite field region following an endothermic feature 4 in DTA curve (Figs. 4, 9). As already mentioned, in the products of A170-category the impurity phases containing nitrogen precipitated in small quantities at the milling stage: fcc-γ and (Fe,Cr)4N for C38–C55 (Fig. 1) and fcc-CrN for A113–A170 [11]. In the products of B170-category, the impurity phases containing nitrogen precipitated/coarsened at the earlier stage of heating, at about 923 K; this process (described in more details in [11]) is consistent with an exothermic feature 1 of energy consumption in DTA registered at the same temperature (Fig. 9a). In both A170- and B170-categories, the precipitated phases containing nitrogen are stable in the ferrite phase field and decomposed by degassing of N2 in the austenite field region. The as-milled samples of B170-category show a single bcc-α phase in XRD (Fig. 1, see also [11]). C3–C24 show a single bcc-α phase even thought they were milled in air in a high rate regime of contamination with nitrogen, d(N)/dt = 0.042 mass% h−1 (Fig. 5). Indeed, upon heating the TG mass loss related to N2 degassing for up to 38 h milled powders of batch C was up to 1.9 mass% (Fig. 5). Therefore, XRD failed to detect the impurity phases containing up to 1.9 mass% nitrogen in powders milled in air up to 38 h. The lack of reflections for contaminants in XRD for 6–24 h milled powders of batch C can be supposed due to the fact that the process of short-time milling with relatively high quantities of nitrogen and oxygen [and thus in a high fracture rate regime (Fig. 3)] maintains these elements adsorbed on the particle surface, in the interstitial sites or at most in the compounds in the incipient stage of precipitation (e.g. nucleation of nitrides [11], the local formation of nanophases with fcc [4] and/or bct [8, 9] structure) that were difficult to detect by XRD (Fig. 1).
The nitrogen degassing (in the studied here temperature range, up to 1373 K) is supported by MS analysis performed on A170 and C170 products as shown in Fig. 9b. A pronounced signal caused by mass 28 (N2) was observed for both samples. The m/z 28 peak was registered at ~ 928 K for C170 and at ~ 1293 K for A170, thus, in the regions were the substantial TG mass losses occur (Fig. 9b). Besides m/z 28, a signal caused by mass 44 (CO2) was registered (not shown) for both samples. The intensity of m/z 44 (CO2) peak, however, is more than one order of magnitude lower than m/z 28 (N2) peak. Moreover, the m/z 44 (CO2) peak was registered above the (studied here) temperature 1373 K where the (Fe,Cr)2O3 to (Fe,Cr)3O4 reduction complete (Fig. 7, see also Fig. 9 in ref [11]); most probably CO2 is a product of carbothermal reaction accompanying the oxide reduction [23, 24]. (During long-term milling the powders might be contaminated with tiny amount of carbon from the container walls/balls). As for the least contaminated samples of batch B milled in Ar without the interruption of milling process, the incorporation of up to 0.5 mass% nitrogen is supported as well by LECO measurements [an amount of 0.496 ± 0.0013 mass% nitrogen was detected in the powder milled for 100 h in good agreement with the total TG mass loss of B170 at temperatures below 1373 K (Fig. 9b)].
It is evident from Figs. 8 and 9 that the product C170 milled in air with interruption of milling process is the most contaminated, whereas the product B170 milled under an Ar atmosphere without interruption of milling process is the least contaminated one. The only proof of the presence of some contaminants in B170 is an insignificant TG mass loss, 0.45 mass%, at about 1313 K (Fig. 9b). The contamination of the batch C powders milled in air was of course expected. However, the contamination of batch A powders has occurred even if they were (re)loaded under an argon atmosphere. The reasons of—and the process of—contamination of batch A powders milled under an Ar atmosphere with interruptions were described in ref [11]. (Briefly from [11]. Since the mill is operated under ambient conditions contamination with nitrogen and oxygen is caused by leak formation and the leakage of air through the seals into the container during milling. A way to minimise the contamination from the air would be to create and maintain a vacuum inside the container during milling. As concluded in [11] this happens if more oxygen and nitrogen is consumed [3] from the milling container at the initial stage of milling. The rate of the oxygen and nitrogen consumption, particularly when the air begins to leak into vial, and thus, the rate of vacuum creation depends on the rate of the reaction of the powder with nitrogen and oxygen. The rate of the reaction of the powder with nitrogen and oxygen depends on the fracture rate of the powder particles determined as well by alloying degree. The high fracture rate implies the high ratio of the total high-reactive surface area to the volume of the particles. If the rate of the absorption of nitrogen and oxygen is higher as compared to the rate of the entering of air into the vial then the pressure in the vial decreases with a higher rate. The created vacuum keeps the vials (air) tight. If the milling process is interrupted after the steady state of Fe–Cr alloying was reached then after resealing and restarting the milling, the fracture rate, and thus the rate of nitrogen and oxygen adsorption, is not as high as that in the initial stage of milling. So, the rate of the reduction of the pressure in the vial is low and does not result in the vacuum creation. This allows the outside atmosphere to continuously leak into the vial during milling and contaminate the powders with nitrogen and oxygen). In addition to the above-described process of vacuum creation at the earlier stage of milling [11] another approach for the conservation of the vacuum and/or of the initial Ar pressure for very long duration of milling can be considered as follows. At the early stage of milling, when plastic deformation and rate of fracture are high (Fig. 3b), the ultrathin and/or ultrafine particles with still low hardness can fill the leaks (caused by the magnitude of the pressure difference across the seal or by the imperfection of integrity of the vial seal [3 and references therein]). This does not allow the outside atmosphere to leak into the vial during milling and keeps the initial Ar pressure or the vacuum created at the earlier stage of milling. This can explain the insignificant contamination of powder B170 loaded in an Ar atmosphere in spite of very long duration of milling in a mill operating under atmospheric conditions. At the moment of interruption of milling and opening the container, the seal of ultrafine particles is destroyed; after reloading and restarting the milling it cannot be recovered especially if the steady state of Fe–Cr alloying was already reached and the rate of fracture is not as high as that in the initial stage of milling. The high alloying degree and the low rate of fracture associated with the low number of ultrathin/ultrafine particles and with the increased particle hardness makes the powder particles unable to fill the leaks and thus to create vacuum or to keep the initial Ar pressure.
Summary and conclusions
Fe–14Cr–3W–0.4Ti–0.25Y2O3 (Fe14Cr) ferritic steel powders loaded in air, milled up to 170 h with interruptions of the milling process and heated up to 1373 K were investigated by thermal analysis in correlation with X-ray diffraction and scanning electron microscopy. The large amounts of impurity phases in the powders milled over 55 h in air, readily detected by XRD, made it possible to understand and to interpret the impurification process and to generalise it for any as-milled powders obtained in any milling conditions irrespective of the milling atmosphere, duration and thus, of amounts of contaminants. The main results of this study are summarised as follows.
-
1.
In the first 6 h of milling in air, the rapid increase in microstrain and lattice constant and, decrease in crystallite size was attributed to the creation and accumulation of defects and to the mixing between the alloying elements.
-
2.
By milling between 6 and 55 h, the microstructure modification (with slower rate) was ascribed to the additional defects generated by nitrogen and oxygen uptake from the air during milling.
-
3.
By milling in air over 55 h, the lattice parameter and the microstrain increase significantly, whereas an ultrafine crystallite size was reached, 4.1 nm, for 170 h milling. The modification of microstructure parameters was ascribed to the supersaturation of the ferrite phase with nitrogen.
-
4.
(Fe,Cr)4N, fcc-γ, (Fe,Cr)2O3 impurity phases were readily detected by XRD in powders milled over 38 h in air.
-
5.
Two regions of mass loss in TG were observed upon heating (depending on milling parameters or milling atmosphere). The TG mass loss below 1093 K in the ferrite phase field was caused by nitrogen degassing after the decomposition of metastable phases formed during milling. The TG mass loss above 1093 K in the austenite phase field was caused by decomposition of residual phases containing nitrogen which are stable in the ferrite phase field, namely the phases that have precipitated in small quantities during milling or have precipitated/stabilised upon heating.
-
6.
At 55 h of milling, the rate of nitrogen uptake from the air changes from fast (d(N)/dt = 0.042 mass% h−1) to slow (d(N)/dt = 0.0132 mass% h−1) regime.
-
7.
A high rate regime of contamination with nitrogen (up to 1.9 mass%) was registered by TG measurements in powders milled up to 38 h although the XRD did not detect the impurity phases containing nitrogen.
-
8.
The contamination of powder milled for 170 h under an argon atmosphere without interruption of the milling process was insignificant (corresponding to less than 0.5 mass% mass loss in TG) as compared to the powders milled in air and under an Ar atmosphere with interruption of milling process.
-
9.
The milling without the interruption of the milling process is an approach for the conservation of the vacuum (created at the earlier stage of milling) and/or of the initial Ar pressure in the containers for very long milling times. The ultrathin/ultrafine particles capable to fill the container leaks were hypothesised to be the impediment against contamination from air.
References
Odette GR. Recent progress in developing and qualifying nanostructured ferritic alloys for advanced fission and fusion applications. JOM. 2014;66:2427–41.
Stork D, Agostini P, Boutard JL, Buckthorpe D, Diegele E, Dudarev SL, English C, Federici G, Gilbert MR, Gonzalez S, Ibarra A, Linsmeier Ch, Puma Li A, Marbach G, Morris PF, Packer LW, Raj B, Rieth M, Tran MQ, Ward DJ, Zinkle SJ. Developing structural, high-heat flux and plasma facing materials for a near-term DEMO fusion power plant: the EU assessment. J Nucl Mater. 2014;455:277–91.
Suryanarayana C. Mechanical alloying and milling. New York: Marcel Dekker; 2004.
Aufrecht J, Leineweber A, Foct J, Mittemeijer EJ. The structure of nitrogen-supersaturated ferrite produced by ball milling. Philos Mag. 2008;88:1835.
Hryha E, Nyborg L. Thermogravimetry study of the effectiveness of different reducing agents during sintering of Cr-prealloyed PM steels. J Therm Anal Calorim. 2014;118:825–34.
HrubovIa´kova´ M, Dudrova´ E, Hryha E, Kaba´tova´ M, Harvanova´J. Parameters controlling the oxide reduction during sintering of chromium prealloyed steel. Hindawi Publishing Corporation Advances in Materials Science and Engineering; 2013, p. 16. Article ID 789373.
Danninger H, Gierl C, Kremel S, Leitner G, Jaenicke-Roessler K, Yu Y. Degassing and deoxidation processes during sintering of unalloyed and alloyed PM steels. Powder Metall Prog. 2002;2(3):125–40.
Rawers JC, Krabbe R, Cook DC, Kim TH. Differences in the microstructure of iron mechanically processed powder alloyed with interstitial and substitutional elements. Nanostruct Mater. 1997;9:145.
Rawers JC, Cook DC. Influence of attrition milling on nano-grain boundaries. Nanostruct Mater. 1999;11:331.
Teus SM, Mazanko VF, Olive JM, Gavriljuk VG. Grain boundary migration of substitutional and interstitial atoms in α-iron. Acta Mater. 2014;69:105.
Mihalache V. Thermal analysis of ball-milled Fe–14Cr–3W–0.4Ti–0.25Y2O3 ferritic steel powder. Evidence for contamination from the air. J Therm Anal Calorim. 2016;124:1179.
Lemoine C, Fnidiki A, Lemarchand D, Teillet J. Grain core study of Fe1-xCrx nanograins obtained by mechanical alloying. J Phys Condens Matter. 1999;11:8341.
Fnidiki A, Lemoine C, Teillet JJ. Structural and magnetic properties of grain boundaries in Fe60Cr40 alloy synthesized by mechanical alloying. J Phys Condens Matter. 2002;14:7221.
Alleg S, Souilah S, Suñol JJ. Thermal stability of the nanostructured powder mixtures prepared by mechanical alloying. In: Elkordy AA, editor. Applications of calorimetry in a wide context—differential scanning calorimetry, isothermal titration calorimetry and icrocalorimetry. Vienna: InTech Publisher; 2013. p. 21–48.
Azzaza S, Alleg S, Sunol JJ. Microstructure characterization and thermal stability of the ball milled iron powders. J Therm Anal Calorim. 2015;119(2):1037–46.
Salahinejad E, Amini R, Hadianfard MJ. Structural evolution during mechanical alloying of stainless steels under nitrogen. Powder Technol. 2012;215:247.
Rawers JC, Maurice D. Understanding mechanical infusion of nitrogen into iron powders. Acta Metall Mater. 1995;43(11):4101–7.
Hebda M, Gądek S, Skałon M, Kazior J. Effect of mechanical alloying and annealing on the sintering behaviour of AstaloyCrL powders with SiC and carbon addition. J Therm Anal Calorim. 2013;113:395–403.
Tavares SSM, Fruchart D, Miraglia S, Laborie D. Magnetic properties of an AISI 420 martensitic stainless steel. J Alloys Compd. 2000;312:307.
Chasoglou D, Hryha E, Nyborg L. Effect of process parameters on surface oxides on chromium-alloyed steel powder during sintering. Mater Chem Phys. 2013;138(138):405–15.
Hryha E, Nyborg L. Oxide transformation in Cr–Mn-prealloyed sintered steels: thermodynamic and kinetic aspects. Metall Mater Trans A. 2014;45:1736–47.
Brust S. Transformation of surface oxides during vacuum heat treatment of a powder metallurgical hot work tool steel. Diploma work; 2013.
Calderon RO, Gierl-Mayer C, Danninger H. Application of thermal analysis techniques to study the oxidation/reduction phenomena during sintering of steels containing oxygen sensitive alloying elements. J Therm Anal Calorim. 2017;127:91–105.
Kirakosyan H, MinasyanT Niazyan O, Aydinyan S, Kharatyan S. DTA/TG study of CuO and MoO3 co-reduction by combined Mg/C reducers. J Therm Anal Calorim. 2016;123:35.
Acknowledgements
This work was supported by the European Community in the framework of the European Fusion Development Agreement (EFDA) under Project WP13-MAT-01-ODSFS-01. The views and opinions expressed in this paper do not necessarily reflect those of the European Commission. The Romanian Ministry of Research is acknowledged for the use of their National Institute of Materials Physics facility under the Core Program: Project PN16480103.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Mihalache, V., Mercioniu, I., Aldica, G. et al. Thermal analysis, microstructure and impurity phases evolution in Fe14Cr ferritic steel powders ball-milled in air and under an argon atmosphere. J Therm Anal Calorim 134, 463–474 (2018). https://doi.org/10.1007/s10973-018-7274-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-018-7274-z