
Abstract
Calcium copper titanate powders were synthesized by a soft chemistry route, aiming to establish a cost-effective solution method to obtain sintered ceramics with giant electric permittivity (ε′) and low dissipation factor (tanδ). Powders and sintered pellets were characterized by several techniques. The thermal decomposition behavior of the porous foam evidences that a hydroxycitrate was formed below 200 °C. Single cubic perovskite-type phase was obtained after calcination of the precursor powder at 700 °C for 5 h. Negligible mass loss occurs above 400 °C. During heating the precursor material, CuO is the first crystallized phase. A giant ε′ and low tanδ are obtained after sintering. The extension of the thermal window of ε′ is wider than those of powders prepared by other methods.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polycrystalline CaCu3Ti4O12 (CCTO) compound with body-centered cubic perovskite-type structure has been thoroughly investigated over near two decades, because of its unusual dielectric properties [1,2,3]. A giant electric permittivity (ε′ > 1000) coupled to a low dissipation factor (tanδ < 1) foreseen technological application of CCTO in microelectronic devices, especially those of low frequency such as cell phones and personal computers [2, 3].
Since the pioneering work of Subramanian et al. [1], the microstructure of this compound has been claimed to play a role on its dielectric properties, and many efforts have been given to correlate the bulk properties of sintered CCTO with microstructure features, like the grain size [3]. Typically, CCTO powder mixtures are prepared by the conventional method of mixing the starting oxides and/or carbonates followed by solid-state reaction (SSR) at high temperatures for relatively long holding times (> 10 h). Nevertheless, such sintering profile may induce deviation from stoichiometry arising secondary phases and promoting exaggerated grain growth. In addition, it was reported that copper stoichiometry and its oxidation state influence the electric permittivity and overall electric response of CCTO, respectively [4, 5].
An approach to overcome these constraints is the use of a soft chemistry method to synthesize this compound. In general, these methods allow for obtaining fine-grained powders with improved homogeneity and high sinterability. Several methods have been used to synthesize CCTO such as combustion [6], simultaneous precipitation [7,8,9], molten salts [10], and variants of the sol–gel method [11,12,13,14,15,16,17,18].
In particular, those methods involving polymeric or gelatinous matrices are known to produce nanosized powders with homogenous elemental distribution, because of the slow rates for cation diffusion in these media. Therefore, these methods are preferred to synthesize complex compositions.
In this work, CCTO was synthesized by a sol–gel soft chemistry route, also known as cation complexation (CC), using citric acid as complexant agent. This method is suitable for obtaining a fine particulate material with narrow size distribution and high sinterability [19]. The properties of powder particles and sintered compacts were evaluated, aiming to obtain an optimized route for the preparation of this dielectric material.
Experimental procedure
Materials and sample preparation
CCTO compound was synthesized by the CC route using reagent grades calcium nitrate tetrahydrate, Ca(NO3)2·4H2O, copper nitrate trihydrate, Cu(NO3)2·3H2O, and titanium isopropoxide, [CH3(CH2)3]4Ti. Anhydrous citric acid, C6H8O7, was employed as complexant agent.
Stock solutions of calcium and copper nitrates were prepared with distilled water. The concentration (1 mol L−1) of the stock solutions was determined by gravimetry. Stoichiometric amounts of the stock solutions were mixed and homogenized in a 250-mL beaker. In parallel, an aqueous solution of citric acid (1:1) was prepared. Titanium isopropoxide was added dropwise to the citric acid solution to form titanium citrate. Afterward, the solution containing calcium and copper nitrates was slowly added to the titanium citrate solution to avoid precipitation. The ratio total metal/citric acid was fixed at 1:1.25. The transparent solution containing the desired cations remained under vigorous stirring and the temperature was raised to 70 °C. The solution gradually turned more viscous with evaporation of water and NOx vapor, and finally very porous blue foam, hereafter named precursor, was obtained. This foam was pre-calcined at 250 °C for 1 h for partial elimination of organic material and subsequently calcined in the 700–900 °C range for 5 and 10 h in static air.
CCTO was also prepared by the conventional solid-state reaction (SSR) method for comparison purpose. In this case, CaCO3 (P.A.), CuO (99.7%), and TiO2 (99.5%) were the starting materials. Stoichiometric amounts of the starting powders were wet milled using isopropyl alcohol and zirconia milling media (ϕ 2 mm) in an attrition mill for 30 min. The powder mixture was dried at 100 °C and deagglomerated in an agate mortar. The calcination was performed at 900 and 950 °C for 10 h in static air. An organic binder (polyvinyl alcohol, molecular weight = 100,000) was added (2 mass%) to the calcined powder prepared by the SSR method to increase the mechanical strength of green compacts.
Cylindrical compacts of CCTO obtained by both methods were prepared by uniaxial pressing (ϕ 9 mm) in stainless steel dye at a pressure of 55 MPa. Sintering experiments were conducted in a resistive furnace (Lindberg BlueM) in powder bed at dwell temperatures of 1070–1090 °C and holding time of 12 h with heating and cooling rate of 10 °C min−1.
Characterization methods
The thermal decomposition of CCTO prepared by soft chemistry route was studied by simultaneous thermogravimetry, TG, and differential thermal analyses, DTA (Netzsch, STA 409) from room temperature up to 1000 °C with heating and cooling rate of 10 °C min−1 in synthetic air and using α-alumina as reference material. Identification of crystalline phases in powders and sintered specimens was accomplished by X-ray diffraction, XRD (Bruker-AXS, D8 Advance) in the 20° ≤ 2θ ≤ 80° with 0.05° step size and 3 s counting time utilizing Ni filtered Cu Kα radiation (λ = 1.5405 Å). The residual carbon content was determined by gas chromatography (Leco, CS400).
The density of sintered compacts was determined by the immersion method with distilled water. Microstructure observations were performed in powder materials and sintered specimens by field emission gun scanning electron microscopy, FEG-SEM (FEI, Inspect F50). The electric and dielectric properties of sintered CCTO specimens were investigated by impedance spectroscopy (Hewlett Packard, 4192A) in the 5 Hz–13 MHz frequency range with 100 mV of applied signal from room temperature (~ 20 °C) up to 180 °C. All specimens remained in an oven at 100 °C overnight before impedance spectroscopy measurements. Silver was used as electrode material.
Results and discussion
Powder characterization and microstructure
Figure 1 shows TG and DTA curves of the precursor material synthesized by the CC route. The endothermic event in DTA curve at ~ 100 °C is obviously related to the elimination of physisorbed water. The exothermic peak at 154 °C is due to thermal decomposition of free citric acid, which was added in excess to the cation solution. The exothermic event at 315 °C accompanied by mass loss is attributed to thermal decomposition of carbon compounds. The mass loss occurs in two stages (I: ~ 100–180 °C and II: ~ 200–400 °C) and is negligible for temperatures higher than 400 °C. This behavior is typical of this method, whenever citric acid is used as complexant agent. The product formed after the first stage of mass loss is a mixed hydroxycitrate [20].

TG-DTA curves in air of the as-synthesized porous foam
TG and DTA curves of the pre-calcined material are shown in Fig. 2. The mass loss observed in the TG curve occurs in two stages: ~ 50–180 and ~ 200–420 °C. Total mass loss amounts 52%. This result corroborates previous report on the high hygroscopic character of the pre-calcined material [21]. The DTA curve shows an endothermic event at ~ 95 °C and exothermic events at ~ 360, 640, and 725 °C. The endothermic peak correlates with that detected in the precursor material (Fig. 1).

TG-DTA curves in air of the pre-calcined precursor material
In order to determine precisely the origin of the exothermic events, small amounts of the pre-calcined material were heated up to 360, 640, 725, and 1000 °C without holding time, and analyzed by XRD (Fig. 3).

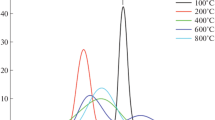
Room-temperature XRD patterns of the pre-calcined material heated up to selected temperatures without holding time
The pre-calcined material heated up to 360 °C is amorphous to X-rays. Then, the exothermic peak at that temperature coupled to a large fraction of mass loss may be assigned to the elimination of organic material. After heating the pre-calcined powder up to 640 °C the XRD pattern consists of only two diffraction peaks at 2θ ~ 37 and 38.7° due to CuO (ICDD 2-1040). However, most of the material is amorphous. This result shows that the cubic perovskite phase of CCTO has not been formed at that temperature and, in addition, that CuO phase is the first one to crystallize. The XRD pattern of the powder heated up to 725 °C shows the most intense diffraction peak of cubic CCTO along with those of CuO. Nevertheless, the crystallinity of the powder is still lower. These results are in general agreement with those previously reported, where single-phase CCTO was obtained for calcination temperatures above 700 °C [6, 10, 13, 15, 17], and mostly 800 °C for precipitated powders [7,8,9]. Finally, after heating the pre-calcined material up to 1000 °C, the XRD pattern corresponds to that of CCTO (ICDD 75-1149) with no traces of other crystalline phases. These finds evidence the relatively low kinetics of CCTO phase formation requiring relatively high temperature and/or long times even for powders prepared by soft chemistry methods.
Figure 4a shows powder XRD patterns of the precursor material after calcination in the 700–900 °C range for 5 and 10 h. All diffraction patterns are single phase with high crystallinity. In contrast, CCTO powder mixture prepared by the SSR method (Fig. 4b) exhibits the cubic CCTO as the main characteristic phase along with minor amounts of CuO and CaTiO3 secondary phases.

Room-temperature XRD patterns after calcinations of the a precursor material and b powder mixture prepared by the SSR method
Nearly all sol–gel-like methods of synthesis give rise to a non-negligible amount of carbonaceous material, which may not be easily eliminated during calcination and hinder or modify in some way the sintering process. Moreover, excess carbon materials may react with calcium generating secondary phases and resulting in deviation from stoichiometry. Then, to optimize the calcination step of the synthesized powder, the carbon content after calcination was determined by gas chromatography (Table 1).
The residual carbon content decreases with increasing the temperature of calcination, as expected, and after 900 °C it amounts about half of the value determined for powders calcined at 700 °C. Therefore, hereafter all experiments were carried out with synthesized CCTO powder calcined at 900 °C for 10 h. The same calcination profile was adopted for the powder mixture prepared by the SSR method.
Figure 5 shows FEG-SEM micrographs of CCTO powders prepared by the (a) CC and (b) SSR methods. The nanostructured powder prepared by the soft chemistry route after calcination at 900 °C does not exhibit microstructure features characteristic of a loosely-bounded powder, but instead it shows necks between small particles revealing that the sintering process has already started. Powders prepared by the SSR method show some small sized particles (non-sintered material) and grains (sintered material). This dual-mode microstructure may be a consequence of non-homogeneous distribution of the starting components and, probably, constitutes the main reason for the frequently observed bimodal distribution of grains for CCTO prepared by the SSR method [22].

FEG-SEM micrographs of calcined CCTO powders prepared by the a CC and b SSR methods
Table 2 lists values of apparent density of specimens prepared by CC and SSR methods. The sintered density is high for specimens prepared by both methods, except for the lower temperature of sintering, and achieves 98% of the theoretical value (4.90 g cm−3, [22]) for specimens prepared by the CC method sintered at 1090 °C. It is worth noting that these specimens did not contain any additive to assist densification.
Figure 6 shows XRD patterns of sintered specimens.

Room-temperature XRD patterns of CCTO sintered specimens
The XRD patterns of CCTO specimens show the main diffraction peaks of the characteristic body-centered cubic phase. Small-intensity peaks of CuO were detected in all specimens. The CCTO specimen prepared by the SSR method exhibits, in addition, the high-intensity peaks of CaTiO3 as impurity phase. Then, this result evidences that specimens prepared by the CC method contain high degree of purity.
FEG-SEM micrographs of fractured surfaces of CCTO prepared by the two methods are depicted in Fig. 7. The specimen prepared by the SSR method exhibits coexisting large (~ 20 μm) and small (less than 1 μm) grains. The residual porosity is most confined at the grain boundaries. The specimen prepared by the CC method shows grains with polygonal shape and with grain sizes of the order of 10 μm along with comparatively narrow distribution in size.

FEG-SEM micrographs of CCTO sintered at 1090 °C
Electric and dielectric properties
Figure 8 shows room-temperature (~ 23 °C) impedance spectroscopy spectra of CCTO pellets sintered at several temperatures and prepared with powders obtained by the CC method. For comparison purpose, the impedance spectroscopy spectrum of CCTO sintered at 1090 °C prepared by the SSR method is also shown.

Impedance spectroscopy spectra of sintered CCTO measured at room temperature (~ 23 °C)
The semicircular arc at lower frequencies (~ 1000 Hz) corresponds to the electrical response (capacitive and resistive effects) of the grain boundaries, with typical capacitance value of the order of nF. The resistivity of grain boundaries is determined from the diameter of the semicircular arc. The electrical response of the bulk (grains) obtained at higher frequencies (≥ 105 Hz) is highlighted in the inset. The grain resistivity is obtained from the nonzero intercept on the Z′ axis.
The analysis of the impedance spectroscopy spectra collected at several temperatures allowed for obtaining the temperature dependence of the electrical conductivity of CCTO (Fig. 9).

Temperature dependence of the electrical conductivity of grains and grain boundaries of CCTO
In the limited temperature range of measurements the electrical conductivity of CCTO may be represented by single straight lines for both grains and grain boundaries. Slight different slopes in these plots are due to changes occurring during sintering, and seem to be related to the copper valence state [23].
Activation energy values calculated from these plots are 9.649–19.3 kJ mol−1 (grains) and 57.89–67.54 kJ mol−1 (grain boundaries). The heterogeneous microstructure of CCTO consisting of semiconducting grains and insulating grain boundaries accounts for the observed difference in the activation energy values.
The temperature dependence of ε′ and tanδ at 1000 Hz are shown in Figs. 10a and 10b, respectively, for CCTO specimens prepared by CC and SSR methods.

Room-temperature dependence of (a) ε′ and (b) tanδ on measuring temperature
The room-temperature values of ε′ (9031) and tanδ (0.09) for CCTO prepared by the CC method agree with those of Subramanian et al. [1]. The CCTO specimen prepared by the SSR method exhibits higher values of ε′ (14,069) and tanδ (0.11). It seems that the dissipation factor increases with increasing the electric permittivity. Therefore, for future technological application of CCTO, a compromise between these two dielectric parameters should be established.
It should be remarked that ε′ and tanδ, for CCTO prepared by the CC method, are weakly temperature dependent. Moreover, the extension of the temperature window for ε′ is wider than those found in previous reports [5, 24]. This result is probably related to the homogeneous microstructure of CCTO prepared by this method of synthesis.
Conclusions
Polycrystalline CCTO powder was successfully synthesized by the CC method. Thermal decomposition of the as-synthesized porous foam revealed formation of a hydroxycitrate as intermediate compound. During heating up to 1000 °C the precursor material, CuO crystallizes before formation of CCTO phase, which was first detected at 725 °C. Single-phase CCTO was obtained after calcination at 700 °C for 5 h. The relative density of specimens prepared by the CC method achieves 98% for sintering at 1090 °C, without introduction of any additive to assist densification. The electric permittivity and dissipation factor of CCTO prepared with CC powder are of the same order of magnitude of previous reported values. The extension of the temperature window of ε’ is wider than those for specimens prepared by other methods.
The CC method used here does not require strict control of pH and other synthesis parameters or procedures such as aging, and may be conducted with cost-effective reagents. Moreover, it allows for obtaining a high degree of phase purity after sintering compared to specimens prepared by the SSR method. Therefore, it is an alternative and suitable route to synthesize CCTO powder.
References
Subramanian MA, Li D, Duan N, Reisner BA, Sleight WA. High dielectric constant in ACu3Ti4O12 and ACu3Ti3FeO12 phases. J Solid State Chem. 2000;151:323–5.
Ramirez AP, Subramanian MA, Gardel M, Blumberg G, Li D, Vogt T, Shapiro SM. Giant dielectric constant response in a copper–titanate. Mater Lett. 2000;115:217–20.
Brizé V, Gruener G, Wolfman J, Fatyeyeva K, Tabellout M, Gervais M, Gervais F. Grain size effects on the dielectric constant of CaCu3Ti4O12 ceramics. Mater Sci Eng. 2006;B129:135–8.
Shao SF, Zhang JL, Zheng P, Wang CL. Effect of Cu-stoichiometry on the dielectric and electric properties in CaCu3Ti4O12 ceramics. Solid State Commun. 2007;142:281–6.
Fang T-T, Mei L-T, Ho H-F. Effect of Cu stoichiometry on the microstructures, barrier-layer structures, electrical conduction, dielectric responses and stability of CaCu3Ti4O12. Acta Mater. 2006;54:2867–75.
Liu J, Smith RW, Mei W-N. Synthesis of the giant dielectric constant material CaCu3Ti4O12 by wet-chemistry methods. Chem Mater. 2007;19:6020–4.
Marchin L, Guillemet-Fritsch S, Durand B, Levchenko AA, Navrotsky A. Grain growth-controlled giant permittivity in soft chemistry CaCu3Ti4O12 ceramics. J Am Ceram Soc. 2008;91:485–9.
Zhao Y, Gao R, Su G, Lin H, Wang C, Cheng C. Effect of dispersant on CaCu3Ti4O12 powders synthesized by oxalate co-precipitation method. Mater Lett. 2013;91:187–90.
Porfirio TC, Muccillo ENS. Influence of lithium disilicate addition on the dielectric properties of chemically synthesized CaCu3Ti4O12. J Mater Sci Mater Electron. 2015;26:3970–5.
Wan W, Yang J, Qiu T, Yuan W-X, Liu C, Zhao X. Preparation of giant dielectric CaCu3Ti4O12 ceramics via molten salt method from NaCl flux. Int J Appl Ceram Technol. 2016;13:382–8.
Liu L, Fan H, Fang P, Chen X. Sol-gel derived CaCu3Ti4O12 ceramics: synthesis, characterization and electrical properties. Mater Res Bull. 2008;43:1800–7.
Thomas P, Dwarakanauth K, Varma KBR, Kutty TRN. Synthesis of nanoparticles of the giant dielectric material, CaCu3Ti4O12 from a precursor route. J Therm Anal Calorim. 2009;95:267–72.
Masingboon C, Thongbai P, Maensiri S, Yamwong T. Nanocrystalline CaCu3Ti4O12 powder by PVA sol-gel route: synthesis, characterization and its giant dielectric constant. Appl Phys. 2009;A96:595–602.
Jesurani S, Kanagesan S, Velmurugan R, Thirupathi C, Sivakumar M, Kalaivani T. Nanoparticles of the giant dielectric material, calcium copper titanate from a sol-gel technique. Mater Lett. 2011;65:3305–8.
He Y, Liu T, Xu Y, Zhao J, Du Z. Synthesis of the giant dielectric constant oxide CaCu3Ti4O12 via ethylenediaminetetraacetic acid precursor. Mater Res Bull. 2012;47:1181–4.
Porfirio TC, Muccillo ENS. Dielectric properties of CaCu3Ti4O12 synthesized by different routes. Adv Mater Res. 2014;975:184–8.
Yang Y, Wang X, Liu B. CaCu3Ti4O12 ceramics from different methods: microstructure and dielectric. J Mater Sci Mater Electron. 2014;25:146–51.
Zhang W, Li L, Li P, Chen W. A Novel method to synthesize CaCu3Ti4O12 with acetylacetonate precursor. Mater Lett. 2016;181:71–3.
Muccillo ENS, Rocha RA, Muccillo R. Preparation of Gd2O3-doped ZrO2 by polymeric precursor techniques. Mater Lett. 2002;53:353–8.
Courty PH, Ajot H, Marcilly C. Oxydes mixtes ou en solution solides sous forme trés divisée obtenus par décomposition thermique de précurseurs amorphes. Powder Technol. 1973;7:21–38.
Rocha RA, Muccillo ENS. Physical and chemical properties of nanosized powders of gadolinia-doped ceria prepared by the cation complexation technique. Mater Res Bull. 2003;38:1979–86.
Fang T-T, Liu CP. Evidence of the internal domains for inducing the anomalously high dielectric constant of CaCu3Ti4O12. Chem Mater. 2005;17:5167–71.
Romero JJ, Leret P, Rubio-Marcos F, Quesada A, Fernández JF. Evolution of the intergranular phase during sintering of CaCu3Ti4O12 ceramics. J Eur Ceram Soc. 2010;30:737–42.
Sangwong N, Yamwong T, Thongbai P. Synthesis, characterization and giant dielectric propertied of CaCu3Ti4O12 ceramics prepared by a polyvinyl pyrrolidone-dimethylformamide solution route. J Electroceram. 2013;31:181–8.
Acknowledgements
The authors greatly acknowledge the financial supports of FAPESP (2013/07296-2), CNPq (304073/2014-8) and CNEN.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Porfirio, T.C., Muccillo, E.N.S. Thermal and electrical properties of CaCu3Ti4O12 synthesized by soft chemistry route. J Therm Anal Calorim 133, 851–857 (2018). https://doi.org/10.1007/s10973-018-7184-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-018-7184-0