
Abstract
A glass with the composition of the SiO2–Na2O–CaO–P2O5 was synthesized by sol–gel method at temperature of 900 °C, and then yttrium-, silver- and cerium-containing glasses were prepared at the same conditions using this base system. The structural and thermal properties of the glass samples were investigated by X-ray diffraction, Fourier transform infrared (FTIR) spectroscopy, scanning electron microscopy, energy-dispersive X-ray (EDX) spectroscopy and differential thermal analysis techniques. The Ca5(PO4)2(SiO4)6 phase having the orthorhombic crystal system is detected for each sample without any secondary phase, and this phase is confirmed by the FTIR spectra. With the addition of Y, Ag and Ce to the SiO2–Na2O–CaO–P2O5 system, the variations in the average crystallite size, crystallinity percent, lattice parameters and unit cell volume are observed. A decrease in the crystallization peak temperature and the changes in the glass transition temperature are seen with the addition of Y, Ag and Ce to the base system. The addition of Y, Ag and Ce to the base glass increases significantly its density. The EDX spectra of the as-prepared samples verify the introduction of the as-mentioned elements.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The SiO2–Na2O–CaO, which is a specific glass matrix, has been produced at different concentrations and temperatures, and the new glasses and glass–ceramics have been manufactured by adding of the different oxides to this base system [1–5]. In 1969, using this ternary system, Hench et al. discovered the 45S5 Bioglass®, which is a major milestone for the clinical applications such as dentistry and orthopedics, with the specific composition of SiO2–Na2O–CaO–P2O5 (45.0 SiO2, 24.5 CaO, 24.5 Na2O, 6.0 P2O5 in mass%), and then this quaternary system has been used as a very popular biomaterial [6–12]. Some studies reported based on this quaternary system can be given as follows. Huang et al. [13] characterized two Na2O–CaO–SiO2–P2O5 glasses based on the bioglasses of 45S5.2 and 55S4.1 and reported these glasses are candidates for the biomedical applications. Taherkhani et al. [14] synthesized the new bioglass samples having the SiO2–CaO–SrO–P2O5 system using sol–gel method and studied the effects of the addition of Sr to the glass structure. Mozafari et al. [15] produced the mesoporous bioactive glass based on SiO2–CaO–P2O5 system and reported its non-toxicity and biocompatibility. Adams et al. [16] studied the SiO2–CaO–Na2O–P2O5 quaternary bioactive glass–ceramic prepared by sol–gel method. The magnetic properties of the bioactive glass having the composition of 41CaO–44SiO2–4P2O5–8Fe2O3–3Na2O (in mol%) were investigated by Shankhwar et al. [17]. The role of Ti addition on structural, biological and antibacterial properties of SiO2–CaO–Na2O–P2O5 glass–ceramics was studied by Riaz et al. [18].
In the present work, the SiO2–Na2O–CaO–P2O5-based glasses were synthesized via sol–gel method. The effects of the additives (e.g., Y, Ag and Ce) on the structural, thermal and morphological properties of this quaternary system were studied by X-ray diffraction (XRD), Fourier transform infrared (FTIR) spectroscopy, scanning electron microscopy, energy-dispersive X-ray (EDX) spectroscopy and differential thermal analysis (DTA) techniques.
Materials and method
Synthesis of the glasses
The chemical compositions (mol%) of the glass samples are given in Table 1. For the preparation of the samples, the sol–gel technique was used. All the chemicals were purchased from Sigma–Aldrich and used without any purification. Tetraethyl orthosilicate (TEOS, Si(OC2H5)4), phosphorus pentoxide (P2O5), sodium carbonate (Na2CO3), calcium nitrate tetrahydrate (Ca(NO3)2·4H2O), yttrium (III) nitrate hexahydrate (Y(NO3)3·6H2O), cerium (III) nitrate hexahydrate (Ce(NO3)3·6H2O) and silver nitrate (AgNO3), respectively, were used as SiO2, P2O5, Na2O, CaO, Y2O3, Ce2O3 and Ag2O sources. For each sample, the mole percentages of SiO2, P2O5 and Na2O were kept at the constant values of 61.80, 5.92 and 5.01, respectively. The synthesis procedures of the glass samples are as follows. Hundred milliliters of 0.1 M nitric acid (HNO3) solution was added into the appropriate amount of TEOS and mixed for 30 min. The as-listed chemicals in Table 1 were added to this solution in sequence from left to right, and then 100 mL of distilled water was added. The final solution was mixed for about 45 min until observing the formation of the xerogel samples. The as-prepared xerogel samples were kept at room temperature for 48 h and dried in an oven at 105 °C for 18 h. Then, the as-dried samples were heated in an electric furnace at 900 °C for 2.5 h and the SiO2–Na2O–CaO–P2O5-based glass powders were observed.
Characterization
For X-ray diffraction (XRD) measurements, the as-synthesized samples were grinded and powdered using an agate mortar. The XRD analyses of the powdered samples were performed using a Rigaku RadB-DMAX II model diffractometer in the 2θ range from 10° to 90° with a step size of 0.02° using Cu Kα (0.15406 nm) with Ni-filtered radiation. Using the KBr pellet method, Fourier transform infrared (FTIR) spectra were performed using a PerkinElmer Spectrum One spectrometer with a spectral resolution of 4 cm−1 in the wave number range from 400 to 4000 cm−1. A Shimadzu DTA 50 was used to investigate thermal behaviors of the as-synthesized glasses. These measurements were taken by heating 10 mg of each sample in a Pt crucible in the temperature range from 25 to 1100 °C at heating rate of 10 °C min−1. The density of the as-produced glasses was calculated from Archimedes method. Each density measurement was repeated three times for each glass sample, and the average values of the density were found. The morphology and elemental analysis of the as-prepared glass samples were performed using a scanning electron microscope (SEM, JEOL JSM 7001F) equipped with an energy-dispersive X-ray (EDX) spectrometer.
Results and discussion
XRD analysis
By analyzing the XRD patterns of the synthesized glass samples depicted in Fig. 1, the as-observed peaks on the XRD patterns are in a perfect harmony with the Joint Committee on Powder Diffraction Standards (JCPDS) reference data for the standard calcium phosphate silicate phase (Ca5(PO4)2(SiO4)6, PDF no: 83–1494) having orthorhombic crystal structure. Delben et al. [19] reported the formation of this phase for the 60SiO2–36CaO–4P2O5 (mol%) glass synthesized by sol–gel emulsion. For estimating the crystallite size (D) of the as-produced glasses, the following well-known Scherrer equation was used [20]
where λ is the X-ray wavelength, which is equal to the value of 0.15406 nm for CuKα radiation, β is the full width at half maximum (FWHM) or integral breadth, and θ is the Bragg angle. The crystallite size is affected by the kind and amount of the additive(s) and is in the range of 7.99–10.33 nm.

XRD patterns of the as-synthesized glasses
Using the values of the total area under crystal peaks (\(\sum {A_{\text{C}} }\)) and total area under amorphous peaks (\(\sum {A_{\text{A}} }\)), the crystallinity percent (X C%) was calculated from the following relation [21]:
As can be seen from Table 2, the X C% values change with the addition of an element (e.g., Y, Ag and Ce), that is, the addition of the as-mentioned elements affects significantly the crystallization mechanism of the SiO2–Na2O–CaO–P2O5 glass-system.
By aid of the Miller indices (hkl) and distance (d) for two adjacent plane, the lattice parameters (a, b and c) of the orthorhombic unit cell were estimated using the following relation [20]:
Then, the unit cell volume for each sample was computed by the following equation.
All the lattice parameters change with the addition of different element(s). With the addition of Y, Ce and Ag, the values of the lattice parameters of a and c of the base glass increase, as well as the unit cell volume (V). The value of the lattice parameter of b also varies, but this change is not a continuous increase or decrease.
FTIR spectroscopy
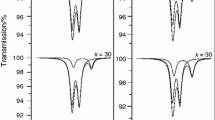
If we look at the FTIR spectra illustrated in Fig. 2, we can see that there is not any notable shift in the positions of the as-observed bands. The assignments of the FTIR bands are summarized as follows. The bands detected at 476 and 726 cm−1 are related to the vibration modes of the Si–O–Si bounds [22, 23]. The band at 799 cm−1 is associated to the Si–O stretching of non-bridging oxygen atoms. The bands observed at 1030 and 1384 cm−1, which are assigned to the silicate network, are interpreted as the Si–O–Si symmetric stretching and the Si–O–Si asymmetric stretching, respectively [14, 24]. The wide band positioned at approximately 1035 cm−1 is ascribed to the stretching vibrations of phosphate groups [19]. Two bands detected at 569 and 603 cm−1 are attributed to the P–O bending vibration mode of the crystalline phosphates [25]. The bands at 3569 and 1637 cm−1 are interpreted as adsorbed water in the samples [26]. We can see that there is not any notable shift in the positions of the as-observed bands, and all the as-observed bands in the FTIR spectra verify the Ca5(PO4)2(SiO4)6 phase detected from the XRD data.

FTIR spectra of the as-produced glass samples
DTA analysis
The exothermic peaks are observed in the DTA plots of the as-prepared glass samples illustrated in Fig. 3. According to the reports in the literature, the peaks appeared on the DTA curves are interpreted as follows. The peak observed in the range of 600–700 °C is associated to the glass transition temperature (T g). The T g values for Pure-G, Ag–G, Ce–G, Y–G and Ag–Ce–Y–G are detected at 639.60, 670.02, 647.45, 609.47 and 681.84 °C, respectively. With the addition of Y to the SiO2–Na2O–CaO–P2O5 glass-system, the value of glass transition temperature decreases, whereas T g value increases with the addition of the rest additives. The exothermic peak located in the interval of 950–1050 °C is attributed to the crystallization of calcium silicates attested from the XRD results [27, 28]. For Pure-G, Ag–G, Ce–G, Y–G and Ag–Ce–Y–G samples, the crystallization peak temperatures (T c) are observed at 1032.83, 965.76, 968.85, 980.17 and 961.27 °C, respectively. The change in T c value can be explained as follows. The addition of new element(s) to the base glass may affect the mobility of the chains of PO4 and SiO4 [29]. It can be seen clearly that all the additives decrease the T c value of the base glass, that is, as expected, the addition of the as-mentioned elements changes the thermal properties of the SiO2–Na2O–CaO–P2O5 glass-system.

DTA curves of the glass samples at the temperature range from 25 to 1100 °C at heating rate of 10 °C min−1
Density measurements
As mentioned previously, the well-known Archimedes method was used to calculate the density (ρ s) of the glasses [30]:
where W a and W b are weights of the sample in air and liquid, respectively. ρ l is the density of the immersion liquid, and the ultra-pure water was used as immersion liquid. According to the reference data for calcium phosphate silicate phase with the PDF no: 83–1494, the theoretical value of ρ s is 2247 kg m−3. The density values are found to be 2263, 2803, 2572, 2317 and 2394 kg m−3 for the Pure-G, Ag–G, Ce–G, Y–G and Ag–Ce–Y–G, respectively. All the additives increase the density of the base glass-system. This increase can be explained as follows. The densities of Ca, Ag, Ce and Y are 1550, 10,490, 6770 and 4472 kg m−3, respectively. The additives of Ag, Ce and Y may substitute with Ca (See Table 1), and the densities of all the additives are significantly greater than that of Ca. The replacement of a lighter element (Ca) with the heavier elements (Ag, Ce and Y) leads to a significant increase in the value of the density. Hence, the increase in the density with the addition of the as-mentioned elements is an expected result.
SEM observations and EDX analysis
Figure 4 shows the SEM images of the as-synthesized glasses, as well as their EDX spectra. From the SEM images, it can be seen that all the additives and their amounts in the glass structure cause the variations in the morphology, and all the samples are composed of the nanoparticles with the different shapes such as rods and/or spheres. The EDX spectra confirm the formation of the calcium phosphate silicate phase for each sample. No impurity is found, and the introduction of the additive(s) is detected. Among the additives, the incorporation of Ce is the highest, while the entrance of Y is the lowest. In brief, the morphology of the base glass is affected significantly by the amount and kind of the additives.

SEM images, EDX spectra and elemental analysis report of the as-obtained glasses
Conclusions
The remarkable consequences of the present study can be sorted as follows. The SiO2–Na2O–CaO–P2O5-based glasses may be produced using the sol–gel route. By analyzing the XRD data, the calcium phosphate silicate phase (Ca5(PO4)2(SiO4)6) with orthorhombic crystal structure is detected for each glass. The amount and kind of the additive(s) affect the lattice parameters, unit cell volume and crystallinity of the samples. From the FTIR spectra, the presence of the functional groups of silicates and phosphates is detected. The findings in FTIR spectra support the XRD results. The changes in the thermal behavior of the base glass in the temperature range of 25–1100 °C are observed from the DTA curves. The glass transition temperature varies, and the crystallization peak temperature decreases with the introduction of the additive(s). Thanks to the element addition, the density of the base glass increases. The EDX results also are in a very good agreement with the XRD and FTIR results.
References
Wakabayashi H, Oishi Y. Liquid-state diffusion of Na2O–CaO–SiO2 system. J Chem Phys. 1978;68:2046–52.
Zhang Z, Xiao Y, Voncken J, Yang Y, Boom R, Wang N, Zou Z. Phase equilibria in the Na2O–CaO–SiO2 system. J Am Ceram Soc. 2011;94:3088–93.
Yao SY, Cao HX, Wang P, Zhang WW, Huo WL, Xu W. Phase transformation and microstructure of wollastonite glass–ceramics in Na2O–CaO–SiO2 system under different heat treatment conditions. Mater Res Innov. 2014;18:657–60.
Kaygili O. Synthesis and characterization of Na2O–CaO–SiO2 glass–ceramic. J Therm Anal Calorim. 2014;117:223–7.
Vitale-Brovarone C, Vernè E, Bosetti M, Appendino P, Cannas M. Microstructural and in vitro characterization of SiO2–Na2O–CaO–MgO glass–ceramic bioactive scaffolds for bone substitutes. J Mater Sci Mater Med. 2005;16:909–17.
Jones JR. Reprint of: review of bioactive glass: from Hench to hybrids. Acta Biomater. 2015;23:53–82.
Hench LL. Bioceramics: from concept to clinic. J Am Ceram Soc. 1991;74:1487–510.
Hench LL. Bioceramics. J Am Ceram Soc. 1998;81:1705–28.
Peitl O, Zanotto ED, Hench LL. Highly bioactive P2O5–Na2O–CaO–SiO2 glass–ceramics. J Non-Cryst Solids. 2001;292:115–26.
Hench LL. The story of bioglass®. J Mater Sci Mater Med. 2006;17:967–78.
Doweidar H. Density–structure correlations in Na2O–CaO–P2O5–SiO2 bioactive glasses. J Non-Cryst Solids. 2009;355:577–80.
Farooq I, Imran Z, Farooq U, Leghari A, Ali H. Bioactive glass: a material for the future. World J Dent. 2012;3:199–201.
Huang LC, Lin CC, Shen P. Crystallization and stoichiometry of crystals in Na2CaSi2O6–P2O5 based bioactive glasses. Mat Sci Eng A Struct. 2007;452–453:326–33.
Taherkhani S, Moztarzadeh F, Mozafari M, Lotfibakhshaiesh N. Sol–gel synthesis and characterization of unexpected rod-like crystal fibers based on SiO2–(1−x)CaO–xSrO–P2O5 dried-gel. J Non-Cryst Solids. 2012;358:342–8.
Mozafari M, Moztarzadeh F, Tahriri M. Investigation of the physico-chemical reactivity of a mesoporous bioactiveSiO2–CaO–P2O5 glass in simulated body fluid. J Non-Cryst Solids. 2010;356:1470–8.
Adams LA, Essien ER, Shaibu RO, Oki A. Sol–gel synthesis of SiO2–CaO–Na2O–P2O5 bioactive glass ceramic from sodium metasilicate. New J Glass Ceram. 2013;3:11–5.
Shankhwar N, Kothiyal GP, Srinivasan A. Understanding the magnetic behavior of heat treated CaO–P2O5–Na2O–Fe2O3–SiO2 bioactive glass using electron paramagnetic resonance studies. Phys B. 2014;448:132–5.
Riaz M, Zia R, Saleemi F, Ahmad R, Hussain T. Influence of titanium on structural, biological and antibacterial properties of SiO2–CaO–Na2O–P2O5 glass–ceramics. Mater Today. 2015;2:5313–7.
Delben JRJ, Pereira K, Oliveira SL, Alencar LDS, Hernandes AC, Delben AAST. Bioactive glass prepared by sol–gel emulsion. J Non-Cryst Solids. 2013;361:119–23.
Cullity BD. Elements of X–ray diffraction. Boston: Addison-Wesley Publishing Company; 1978.
Kaygili O, Tatar C. The investigation of some physical properties and microstructure of Zn-doped hydroxyapatite bioceramics prepared by sol–gel method. J Sol–Gel Sci Technol. 2012;61:296–309.
Catteaux R, Grattepanche-Lebecq I, Désanglois F, Chai F, Hornez JC, Hampshire S, Follet-Houttemane C. Synthesis, characterization and bioactivity of bioglasses in the Na2O–CaO–P2O5–SiO2 system prepared via sol gel processing. Chem Eng Res Des. 2013;91:2420–6.
Majhi MR, Pyare R, Singh SP. Studies on preparation and characterizations of CaO–Na2O–SiO2–P2O5 bioglass ceramics substituted with Li2O, K2O, ZnO, MgO, and B2O3. Int J Sci Eng Res. 2011;2:1–9.
Mozafari M, Moztarzadeh F, Rabiee M, Azami M, Maleknia S, Tahriri M, Moztarzadeh Z, Nezafati N. Development of macroporous nanocomposite scaffolds of gelatin/bioactive glass prepared through layer solvent casting combined with lamination technique for bone tissue engineering. Ceram Int. 2010;36:2431–9.
Catauro M, Dell’Era A, Ciprioti SV. Synthesis, structural, spectroscopic and thermoanalytical study of sol–gel derived SiO2–CaO–P2O5 gel and ceramic materials. Thermochim Acta. 2016;625:20–7.
Dziadek M, Zagrajczuk B, Menaszek E, Wegrzynowicz A, Pawlik J, Cholewa-Kowalska K. Gel-derived SiO2–CaO–P2O5 bioactive glasses and glass-ceramics modified by SrO addition. Ceram Int. 2016;42:5842–57.
Saravanapavan P, Hench LL. Mesoporous calcium silicate glasses. I Synth J Non-Cryst Solids. 2003;318:1–13.
Li HC, Wang DG, Hu JH, Chen CZ. Effect of the partial substitution of K2O, MgO, B2O3 for CaO on crystallization, structure and properties of Na2O–CaO–SiO2–P2O5 system glass–ceramics. Mater Lett. 2013;106:373–6.
Lázaro GS, Santos SC, Resende CX, dos Santos EA. Individual and combined effects of the elements Zn, Mg and Sr on the surface reactivity of a SiO2·CaO·Na2O·P2O5 bioglass system. J Non-Cryst Solids. 2014;386:19–28.
Kaygili O, Yavuz H. The effects of gamma irradiation on non-isothermal crystallization kinetics and microhardness of the Li2O–Al2O3–SiO2 glass–ceramic. J Therm Anal Calorim. 2010;102:681–4.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kaygili, O., Keser, S., Tatar, C. et al. Investigation of the structural and thermal properties of Y, Ag and Ce-assisted SiO2–Na2O–CaO–P2O5-based glasses derived by sol–gel method. J Therm Anal Calorim 128, 765–770 (2017). https://doi.org/10.1007/s10973-016-6012-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-016-6012-7