
Abstract
The multiple melting behavior of many semicrystalline polymers is generally attributed to fusion of original crystals reorganized/recrystallized upon heating at conventional scanning rates. In the present study, the triple and double melting behavior of poly(butylene terephthalate) (PBT) and isotactic polystyrene (iPS), which is observed after isothermal crystallization at low and high T c s, respectively, is put into relation with the presence and absence of rigid amorphous/crystal interphase around the original primary crystals. Such constrained amorphous interphase has been recognized as decisive for the definition of the mechanical, thermal, and barrier properties of semicrystalline polymers. The method for the approximate determination of the limit temperature for the presence of this rigid amorphous fraction (RAF), already applied to poly[(R)-3-hydroxybutyrate] (PHB) and poly(ethylene terephthalate) (PET), is here utilized for PBT and iPS. The complex melting behavior displayed by all these polymers is explained by assuming that two distinct morphologies of primary crystals develop during crystallization at temperatures lower than the RAF limit temperature, in a percentage that is a function of the crystallization temperature. Simultaneously with the RAF devitrification, these distinct morphologies differently reorganize/recrystallize upon heating at low scanning rate, originating the triple melting behavior. On the contrary, after crystallization at high T c s, a single reorganization route is followed by all the primary crystalline structures, characterized by the same morphology made of tightly chain-folded lamellae and absence of vitrified RAF at the amorphous/crystal interphase.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The traditional description of semicrystalline polymers as constituted by two separate phases, amorphous and crystalline, has been replaced by a more accurate one that takes into account also the intermediate nanophase present at the interface between the crystalline and the surrounding amorphous regions [1, 2]. The decoupling between crystalline and amorphous phases is in general incomplete, due to the length of the polymer molecules, which is much higher than the dimensions of the crystalline phase, at least in one direction. The interphase between crystalline and amorphous regions is non-crystalline, has nanometric dimensions, and includes amorphous chain portions whose mobility is hindered by the near crystalline structures. This interphase is generally named “rigid amorphous fraction” (RAF), its mobility being lower than that of the unconstrained amorphous phase, which is usually addressed as “mobile amorphous fraction” (MAF).
A detailed knowledge of the nanoscale phase behavior of semicrystalline polymers is decisive for the assessment of their mechanical, thermal, barrier, and other macroscopic properties [3–12]. The rigid amorphous fraction influences the elastic modulus, with a behavior close to that of the crystal phase [3–7], and affects also the gas barrier properties, due to its higher free volume with respect to the mobile amorphous regions [8–12].
The relationship between crystal growth and rigid amorphous fraction development and between melting and rigid amorphous fraction mobilization has been detailed in the literature for several semicrystalline polymers by utilizing various experimental techniques [13–28]. These studies, performed independently of one another, in different crystallization and melting conditions and with different methods of data processing, if combined and compared with one another, prove that the thermal history defines the combined evolution of crystalline and rigid amorphous phases.
The influence of the crystallization temperature on the RAF development was systematically investigated for poly(l-lactic acid) (PLLA) and poly[(R)-3-hydroxybutyrate] (PHB) [20–22]. Both in PLLA and in PHB, the rigid amorphous fraction was found to develop in parallel to the crystal phase at low crystallization temperatures (T c s), whereas at higher T c s, RAF vitrification was observed only during the final stages of crystallization, in correspondence with the growth of secondary crystals. At T c s higher than 130 °C, RAF was found not to develop at all during isothermal crystallization of PLLA. Full mobilization of the RAF upon heating was reported for both PLLA and PHB at the same temperature at which the RAF starts to develop upon crystallization. Therefore, a limit temperature for the formation and the disappearance of the rigid amorphous fraction in PLLA and PHB was suggested, which resulted, for the investigated grades, approximately 130 and 70 °C, respectively [20–22].
In isotactic polystyrene (iPS) crystallized at 140 and 170 °C, i.e., sufficiently above the glass transition of the MAF (T g = 100 °C), the rigid amorphous fraction increases markedly after spherulite impingement, which corresponds to the beginning of crystallization in restricted regions [17, 18]. On the contrary, upon slow cooling from the melt, with crystallization peak centered at 210 °C, RAF vitrification takes place in iPS during the final stages of the non-isothermal crystallization process, starting from approximately 190 °C, with the full establishment of the rigid amorphous structure completed during the subsequent cooling to room temperature [19].
For poly(ethylene terephthalate) (PET), the temperature at which RAF starts vitrifying upon cooling during non-isothermal crystallization and the temperature at which RAF finishes devitrifying during the successive heating were found approximately coincident [23]. Such value, 215 °C, was proposed as the limit temperature for the presence of RAF in PET, in agreement with other literature studies [16, 29].
The development of RAF during isothermal crystallization was proven also for poly(butylene terephthalate) (PBT): A reduction in the crystallization rate at T c ≤ 170 °C was rationalized as due to RAF formation, which hinders crystallization by the creation of an immobilized amorphous layer around the growing crystals [30].
This limit temperature can be related to the mobility of the polymer chains: At high crystallization temperature, the macromolecules have high mobility, which facilitates the organization of the polymeric segments into ordered crystal structures, with reduced stress transmitted to the amorphous segments, and, in turn, no or minor fraction of amorphous chain segments subjected to geometrical constraints. Conversely, the low chain mobility at low crystallization temperatures implies a more difficult organization of the entangled chain segments into ordered crystal structures, so that a larger rigid amorphous fraction develops.
In the present study, a calorimetric method for the approximate determination of the limit temperature for the presence of RAF in a semicrystalline polymer, recently applied to PHB and PET [22, 23], is implemented to other two common polymers: PBT and iPS. In the previous studies on PHB and PET [22, 23], the RAF limit temperature was put into relation with the corresponding melting behavior. Also for PBT and iPS, in the present study, an analysis of the melting behavior after isothermal and non-isothermal crystallizations is presented and discussed. The aim of these studies is to collect all the experimental evidences up to now available on the identification and quantification of the rigid amorphous/crystal interphase, which could be typical of a large number of polymers.
Both the previously investigated polymers, PHB and PET, and the polymers here studied, PBT and iPS, show a multiple melting behavior, which is a phenomenon very common for semicrystalline polymers. Different reasons have been adduced to explain this behavior, such as the presence of more than one crystalline form [31], molar mass segregation during crystallization or annealing [32, 33], fusion of crystals and confined nanocrystals [34], orientation effects [35], and, more frequently, the occurrence of a melting/recrystallization/remelting process. The multiple melting behavior of PET, PHB, PBT, and iPS is usually associated to the latter mechanism; namely, it has been attributed to the fusion of original lamellae, followed by recrystallization and final fusion of the crystals perfected during heating [36–45], even if a combination of the melting/recrystallization mechanism and a dual distribution of lamellar thickness or lamellar perfection has also been proposed for PET and iPS [46, 47].
Although the mechanism has been identified, a connection between a specific thermal history and the appearance of a double or triple well-resolved endotherm has not yet been sufficiently investigated. A full understanding of the origin of the multiple melting behavior can afford also a new insight into the crystallization process of semicrystalline polymers.
Experimental
Poly(butylene terephthalate) (PBT), with M w = 81,000 g mol−1 and M w/M n = 1.8, purchased from Sigma-Aldrich Corp. in the form of large chips, was compression-molded at 250 °C for 3 min, to obtain films with thickness of about 200 μm. Isotactic polystyrene (iPS), with M w = 556,000 g mol−1 and M w/M n = 1.9, was purchased from Polymer Laboratories (Church Stretton, UK). Being a powder, it was used as received, without further purification or thermal treatment.
Differential scanning calorimetry (DSC) measurements were performed with a Perkin Elmer Differential Scanning Calorimeter DSC 8500 equipped with an IntraCooler III as refrigerating system. The instrument was calibrated in temperature with high purity standards (indium, naphthalene, cyclohexane) according to the procedure for standard DSC [48]. In order to gain precise heat capacity data from the heat flow rate measurements, each scan was accompanied by an empty pan run and calibration with sapphire [48]. Dry nitrogen was used as purge gas at a rate of 30 mL min−1. A fresh sample was employed for each analysis in order to minimize thermal degradation. The sample mass was kept as small as possible, approximately 4 mg, with the aim of minimizing temperature gradients. All the results presented and discussed are the average of three repeated runs. Before the analyses, each PBT and iPS sample was heated at a rate of 50 °C min−1 from room temperature to complete fusion, namely to 250 and 240 °C, respectively, and maintained at this temperature for 3 min [49]. Successively, PBT and iPS were crystallized both non-isothermally and isothermally.
Non-isothermal crystallizations were conducted at −2 °C min−1 from the temperatures of complete fusion down to below T g. At the end of the non-isothermal crystallizations, the samples were reheated at +2 °C min−1 from below T g up to complete melting by conventional DSC and by temperature-modulated DSC (TMDSC), with temperature amplitude (A T) of 1.0 °C and modulation periods (p) of 60 and 120 s. TMDSC analyses were designed using a sawtooth modulation temperature program. The reversing specific heat capacity (c p,rev) was obtained through [50, 51]:
where ω is the frequency of temperature modulation (ω = 2π/p), m the mass of the sample, and A HF(T) the amplitude of the modulated heat flow rate. The frequency-dependent calibration factor, K(ω,T), determined by calibration with sapphire, was 1.05 and 1.00 for p = 60 and 120 s, respectively. The correctness of the calibration factor was proven by the good agreement between the measured solid and liquid specific heat capacities and the literature solid and liquid c p data [52].
Isothermal crystallizations of PBT and iPS were conducted at various temperatures (T c), after ballistic cooling from the temperature of complete fusion, down to T c. PBT was isothermally crystallized at 185 °C ≤ T c ≤ 210 °C for 40 min, and iPS at 150 °C ≤ T c ≤ 210 °C for 120 min and at T c = 220 °C for 240 min, up to completeness of the process, as attested by preliminary measurements. At the end of crystallization, the samples were reheated at +5 and +10 °C min−1 directly from T c up to complete melting.
Results and discussion
Figure 1 shows the experimental specific heat capacity (c p) of PBT during cooling at −2 °C min−1 and the subsequent heating at +2 °C min−1, respectively, measured by conventional DSC. The c p curves reveal that the crystallization process starts at about 205 °C, with peak centered at 200 °C, and a long small tail that extends down to approximately 70 °C, whereas the fusion, which ends at 228 °C, presents a double melting endotherm, with peaks located at 217 and 225 °C. The extension of the crystallization to low temperatures for PBT has been reported, but not interpreted [30]. The DSC plot at +2 °C min−1 is compared in Fig. 2 with the corresponding reversing specific heat capacity curves (c p,rev) by TMDSC, measured at +2 °C min−1, with p = 60 and 120 s. On the same graph, the specific heat capacity data in the liquid and solid state of PBT, as taken from the literature, are also indicated [52]. The fully Y-axis scaled curves are depicted in the inset. The glass transition, connected to the devitrification of the MAF, is centered approximately at 41 °C, in agreement with literature data [53]. Above the glass transition region, the c p and c p,rev curves overlap. At temperatures higher than 75 °C, indicated by a thin arrow, the c p,rev values become frequency dependent, which suggests the occurrence of melting and reorganization of PBT crystals into more stable structures. The amplitude of the modulated heat flow derives from the sum of the absolute values of the endothermic and exothermic events intensity occurring within each modulation period, whereas the latent heat released or absorbed sum algebraically in the specific heat capacity [54, 55]. The marked frequency dependence of c p,rev at temperatures higher than 75 °C proves that, above this temperature, thermal processes involving reversing exchanges of latent heat take place, as typical of polymer melting [2, 13]. In correspondence of the polymer melting, a decrease in modulation frequency leads to a higher c p,rev because a decrease in the frequency of modulation permits a larger percentage of crystalline material to follow the modulation within a single temperature cycle [2, 56]. Therefore, the temperature of 75 °C can be assumed as the starting point of the melting, in agreement with the literature [57].

Specific heat capacity curves (c p) of PBT upon cooling from the melt at −2 °C min−1 (dashed line) and the successive heating at +2 °C min−1 (solid line). The inset shows an enlargement of the c p curves

PBT: enlargement of the specific heat capacity curve (c p, thick solid line) and reversing specific heat capacity curves (c p,rev) with modulation periods of 60 s (thin solid line) and 120 s (thin dashed line) upon heating at +2 °C min−1 after non-isothermal crystallization at −2 °C min−1. The thin dotted lines are the solid and liquid PBT specific heat capacities [52]. The thick dashed line is the approximate linear baseline, whereas the solid line with small circles is the two-phase approximate baseline (c p,base,2phase) calculated by Eq. (3). The inset shows the entire specific heat capacity curves
The variation of crystalline fraction (w C) of PBT upon heating was calculated from the following equation [23]:
where T o is a reference temperature in the melt, Δh °m (T) the temperature evolution of the heat of fusion of the 100 % crystalline polymer, as taken from the literature [52], and c p,base, as a first approximation, the linear baseline constructed from 75 °C up to the end of the melting peak. The linear baseline allows to integrate the c p melting endotherm by Eq. (2) and derive the temperature-dependence of w C, which is presented in Fig. 3 as solid line.

Temperature evolution of crystalline (w C, solid line), mobile amorphous (w MAF, dotted line), and rigid amorphous (w RAF, dashed line) weight fractions of PBT upon heating at +2 °C min−1, after non-isothermal crystallization at −2 °C min−1
From the calculated w C curve, a two-phase approximate baseline heat capacity (c p,base,2phase), which neglects vitrification of rigid amorphous fraction, can be calculated:
where c p,S and c p,L are the thermodynamic specific heat capacities of solid and liquid PBT [52].
Figure 2 shows that c p,base,2phase crosses the linear c p,base at about 190 °C, indicated by a thick arrow. A more accurate baseline can thus be constructed by merging: (1) the linear baseline from 75 °C up to the intersection point, located at 190 °C, and (2) the c p,base,2phase sigmoidal curve, from the intersection point up to the melt. This new baseline leads to a crystalline fraction evolution, determined by Eq. (2), practically coincident with that determined with the linear baseline (∆w C = 0.001 at 50 °C). The overall error of the calculated w C is estimated approximately ±0.01.
The intersection point can be assumed as the approximate limit temperature for the presence of RAF because at higher temperatures all the amorphous segments are mobilized, as the thermodynamic c p is represented by the c p,base,2phase curve, which comprises a crystalline phase and a single amorphous phase. Conversely, at temperatures lower than the intersection point, the thermodynamic c p is lower than the c p,base,2phase curve, which means that below this temperature, part of the amorphous chains are vitrified, being the specific heat capacity of the RAF lower than that of the mobile amorphous phase [58].
The evolution upon heating of the mobile amorphous weight fraction, calculated according to the following relationship [15]:
together with the rigid amorphous weight fraction evolution, obtained by difference, being w C + w MAF + w RAF = 1, is depicted in Fig. 3. The errors for the calculated w MAF and w RAF are approximately ±0.01 and ±0.02, respectively. In the glass transition region, the w RAF curve reproduced in Fig. 3 reflects devitrification of the mobile amorphous fraction. In the temperature range from 50 to 75 °C, the RAF content reduces from about 0.26 to approximately 0.15. This RAF decrease, well evidenced by the increase in the c p and c p,rev values in the temperature range 50–75 °C (Fig. 2), indicates that mobilization of constrained amorphous segments, developed during cooling, starts immediately above the glass transition. At higher temperatures, RAF devitrification goes on progressively up to the complete mobilization.
Similar data treatment was applied to iPS. The c p curves of iPS during cooling at −2 °C min−1 and the subsequent heating +2 °C min−1, measured by conventional DSC, are presented in Fig. 4, whereas the DSC and TMDSC data collected at +2 °C min−1 after non-isothermal crystallization at –2 °C min−1 are reported in Fig. 5. The glass transition of the MAF is centered approximately at 95 °C, in agreement with literature data [52]. The crystallization process at −2 °C min−1 starts at about 235 °C, and its peak is centered at about 209 °C, whereas the fusion, which presents a single melting endotherm with a shoulder on the low-temperature side, has a peak centered at about 227 °C.

Specific heat capacity curves (c p) of iPS upon cooling from the melt at −2 °C min−1 (dashed line) and the successive heating at +2 °C min−1 (solid line). The inset shows an enlargement of the c p curves

iPS: enlargement of the specific heat capacity curve (c p, thick solid line) and reversing specific heat capacity curves (c p,rev) with modulation periods of 60 s (thin solid line) and 120 s (thin dashed line) upon heating at +2 °C min−1 after non-isothermal crystallization at −2 °C min−1. The thin dotted lines are the solid and liquid iPS specific heat capacities [52]. The thick dashed line is the approximate linear baseline, whereas the solid lines with small circles is the two-phase approximate baseline (c p,base,2phase) calculated by Eq. (3). The inset shows the entire specific heat capacity curves
As for PBT, the linear baselines were constructed starting from the temperature, indicated by a thin arrow in Fig. 5, at which the reversing specific heat capacities become frequency dependent. The intersection point between the linear and the two-phase sigmoidal baselines, indicated by a thick arrow in Fig. 5, is located at 194 °C. This value is the approximate limit temperature for the presence of RAF in iPS, or the glass transition temperature of the most constrained portions of RAF. The crystalline, mobile amorphous and rigid amorphous fractions evolution for iPS heated at +2 °C min−1, after cooling at −2 °C min−1, is calculated through Eqs. (2) and (4) after merging, at the intersection point, the linear baseline to the c p,base,2phase sigmoidal curve. The data are illustrated in Fig. 6. At 110 °C, i.e., immediately above the glass transition of the MAF, the RAF content is 0.22. Devitrification of the RAF proceeds progressively up to the final mobilization, which occurs approximately at the beginning of the melting process.

Temperature evolution of crystalline (w C, solid line), mobile amorphous (w MAF, dotted line), and rigid amorphous (w RAF, dashed line) weight fractions of iPS upon heating at +2 °C min−1, after non-isothermal crystallization at −2 °C min−1
As reported in the “Introduction” section, both PBT and iPS display a multiple melting behavior, which is often rationalized as due to a melting/recrystallization/remelting mechanism [39–42], even if a combination of the melting/recrystallization mechanism and a dual distribution of lamellar thickness or lamellar perfection has also been proposed for iPS [47]. Previous analyses of the melting behavior of PHB and PET [22, 23, 59] demonstrated that, for these two polymers, the transition from triple to double melting behavior occurs after isothermal crystallization at the temperatures corresponding to the respective RAF limit temperatures. In particular, investigation on the melting behavior of PET was performed by varying both the crystallization times and the heating rate. The shape of the overall endotherm was found to change, but the number of the melting peaks remained unaltered. For this reason, the triple and double melting behavior was tentatively put into relation with the morphology of the original crystals, and, in particular with the presence and absence of vitrified amorphous interphase around the original primary crystals, respectively.
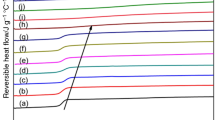
To verify whether this peculiarity is typical also of PBT and iPS, the melting behavior of these two polymers was analyzed by DSC after isothermal crystallization at various temperatures. Figures 7 and 8 collect the c p curves of PBT and iPS, monitored upon heating at two different rates, 5 and 10 °C min−1, in order to disclose the effect of reorganization during heating. Crystallizations were conducted in a wide temperature range, below and above the estimated limit for the presence of RAF. As general feature, for both the analyzed polymers, samples crystallized at low T c s exhibit three endotherms of different relative intensity, whereas samples crystallized at high T c s display two peaks. At the highest investigated T c s, in some cases, a single melting peak can be observed. In Figs. 7 and 8, the endotherms are labelled I, II, and III in order of increasing temperatures.

Specific heat capacity (c p) of PBT upon heating at a 5 °C min−1 and b 10 °C min−1 after isothermal crystallization at T c = 185, 190, 195, 200, 205, and 210 °C for 40 min. The ordinate values refer only to the bottom curve (T c = 185 °C). All the other curves are shifted vertically for the sake of clearness. The thick solid lines are the c p curves after crystallization at 205 °C. The thin solid lines serve as guides for the eyes, to evidence the location of the endothermic events labelled I, II, and III

Specific heat capacity (c p) of iPS upon heating at a 5 °C min−1 and b 10 °C min−1 after isothermal crystallization at T c = 150, 160, 170, 180, 190, 200, and 210 °C for 120 min and 220 °C for 240 min. The ordinate values refer only to the bottom curve (T c = 150 °C). All the other curves are shifted vertically for the sake of clearness. The thick solid lines are the c p curves after crystallization at 200 °C. The thin solid lines serve as guides for the eyes, to evidence the location of the endothermic events labelled I, II, and III
Endotherm I, observed approximately 10–20 °C above T c, has been often associated with the fusion of defective secondary lamellae, which grow in the presence of more perfect crystals during secondary crystallization and melt earlier because of their lower stability [36, 37, 39–41, 43–45]. It has also been attributed to the onset of the overall melting process, being this melting partially masked by the immediate recrystallization [38, 42], and/or to enthalpy recovery subsequent to structural relaxation of the portion of RAF that mobilizes simultaneously with fusion of smaller or less perfect crystals [55]. The second melting peak (II) increases in size and moves to higher temperatures with increasing T c. This endothermic event has been often associated to the fusion of the crystals grown at T c by primary crystallization [36–45].
Conversely, endotherm III, almost independent of T c, has been attributed to melting of the lamellae perfected upon heating [36–39]. The ratio of the area of endotherm III to the area of endotherm II increases with reducing both the heating rate and T c, as shown in Figs. 7 and 8: Upon slow heating more time is available for the recrystallization/perfection of the crystal lamellae, which are increasingly more defective with the decrease of T c, which confirms that a reorganization/perfecting process occurs during the heating scan. As alternative explanation, it has also been proposed that that the multiple melting behavior arises from a multimodal lamellae thickness distribution: Endotherm II originates from the melting of less perfect or thinner lamellae, whereas endotherm III might be due to the melting of more perfect or thicker lamellae, and, to less extent, to the melting of a population recrystallized during heating [45, 46]. Unfortunately, such hypothesis does not rationalize why the more perfect or thicker lamellae, which would give rise to endotherm III, should not develop at high T c s.
It is worth noting that also for the analyzed polymers, as for PHB and PET [22, 23, 59], transition from the triple to the double melting behavior occurs after isothermal crystallization at a temperature (205 and 200 °C for PBT and iPS, respectively), which is close to the RAF limit temperatures, as determined above. This leads to hypothesize also for PBT and iPS a possible influence of the RAF on the respective multiple melting behavior.
The discontinuity in the melting behavior of PBT and iPS, as in the case of the previously studied PHB and PET, can be explained by assuming a distribution of morphologies and/or crystalline perfection as a function of the crystallization temperature. At high T c, the polymer chains have high mobility, which facilitates organization of the polymeric segments into ordered crystal structures, with high percentage of tight adjacent reentry folding and limited stress transmitted to the amorphous segments, which does not lead to vitrification of rigid amorphous fraction. With decreasing T c, the percentage of defective crystalline structures, constituted by small crystalline domains and chain cluster aggregations, increases progressively, accompanied by an increasing fraction of amorphous chain segments subjected, at the crystal boundaries, to geometrical constraints.
In practice, two different morphologies of primary crystals, with similar crystal size, could develop during isothermal crystallization at temperatures lower than the RAF limit temperature, in a proportion that is a function of the crystallization temperature: (1) chain cluster structures and/or crystals with irregular adjacent reentry folding, many loops, and a high percentage of rigid amorphous fraction on the boundaries and (2) crystals with a high percentage of regular adjacent reentry folding and reduced constraints at the amorphous/crystal interphase [59]. The RAF devitrification, which occurs simultaneously with the melting of the less perfect crystals, if it is sufficiently slow, can promote continuous and progressive recrystallization and/or additional crystallization, particularly at the amorphous/crystal boundaries. In particular, rearrangements of the constrained loops could result in an increased crystal thickness and in a higher thermal stability of crystals with larger dimensions, which undergo fusion at higher temperatures, originating endotherm III.
Also the more perfect original crystalline regions, whose fraction increases with T c, constituted by crystals grown with a high percentage of regular adjacent reentry folding and reduced constraints on their boundaries, can melt/recrystallize and reorganize during the heating scan. In this case the positive action exerted by the RAF concerted mobilization is absent, which results in a slightly lower melting temperature (endotherm II), as fusion and recrystallization of chain-folded lamellae is a multistep process that requires the detachment of all the crystalline stems, their diffusion between other amorphous segments, and their successive ordering into a new thicker crystalline structure. The process is expected to be faster in case of smaller lateral dimensions of the lamellae. Thus, the two distinct primary morphologies differently transform upon heating at low scanning rate, originating, together with a population of more defective crystals, the triple melting endotherm. Conversely, after crystallization at T c s higher than the RAF limit temperature, all the primary crystalline structures, which probably are characterized by the same morphology made of tightly chain-folded lamellae and absence of rigid amorphous fraction, undergo the same reorganization route upon heating, originating, together with a population of more defective crystals, the double melting endotherm, with full disappearance of endotherm III. The reorganization process during heating could involve crystals perfection through the annihilation of defects inside the lamellae or at the interfaces of the crystalline regions.
The fusion process of iPS, as well as of PET, was studied also at very high scanning rates, up to 1000 °C s−1, after crystallization at various T c s [38, 42]. At high heating rates, the times allowed for reorganization dramatically reduces, which hinders the recrystallization and the rearrangements of the constrained amorphous segments at the interface. As result, also after isothermal crystallization at low temperature, only one broad melting peak, connected to the fusion of the original, non-reorganized crystals, is detected. Conversely, low heating rates allow a larger extent of chain reorganization involving crystals and the amorphous/crystal interphase, which occurs at timescale typical of conventional DSC. A small variation of the heating rates induces changes in the relative intensities of endotherms II and III, giving indications on the original crystalline morphologies of the as-crystallized polymers. In addition, according to the studies on iPS and PET performed at very high scanning rates [38, 42], the presence of a single melting peak would show that a broad monomodal distribution in crystal size and crystal stability develops in isothermal conditions. Actually, in the case of a polymer crystallized isothermally, containing crystals with similar lamellar thickness, a single wide melting peak, which may not be resolved at very high heating rates, could mask the fusion of crystals with different morphologies, i.e., with different thermal stability.
A further confirmation of the effect of the crystallization temperature on the melting behavior is provided by the non-isothermal crystallization experiments. The non-isothermal crystallization of PBT occurs at temperatures lower than 205 °C, at which RAF should develop (see Fig. 1), and correspondingly, the fusion displays a more complex behavior. Conversely, the non-isothermal crystallization of iPS occurs mainly at temperatures higher than 200 °C, without RAF development (see Fig. 2), which leads to a fusion profile with a smaller number of peaks.
An analysis of the lamellar crystal thickness from SAXS data revealed for PET the occurrence of two lamellar stack populations in samples crystallized at low temperatures (T c < 190 °C), whereas for samples crystallized at higher temperatures, a population of lamellar stacks with a monomodal thickness distribution was evidenced [60]. In parallel, a study on the nanostructural reorganization process occurring in PET samples crystallized at T c lower than 190 °C, i.e., at temperatures at which RAF should develop, revealed an extensive structural reorganization during heating. This reorganization was found to produce the appearance of new lamellar stacks, which contribute to the high-temperature endotherm [61]. These results confirm our suggestion that in PET the RAF, which devitrifies progressively during heating and is completely devitrified approximately at 215 °C, influences the rearrangements of the crystals. Mobilization of the RAF can favor and promote recrystallization of a crystalline population with a higher melting temperature [23]. A further validation of this thesis arises from small-angle X-ray scattering and wide-angle X-ray diffraction studies, performed on PET samples heated and annealed above the crystallization temperatures. These analyses pointed out a change in structural parameters around 215 °C [62, 63]. A discontinuity in the reorganization mechanism was conjectured: Fontaine et al. [62] hypothesized that crystal reorganization during annealing occurs via crystal perfection at the amorphous/crystals boundaries above 215 °C, and Jonas et al. [63] found that, upon heating, the average thickness of the crystalline lamellae increases markedly and dramatically above 215 °C.
As regards PBT, the recrystallization rate was found approximately constant at temperatures lower than 210 °C, whereas a decrease in the recrystallization rate at temperatures higher than 210 °C was reported [64]. This experimental result is in very good agreement with our finding, i.e., that at temperatures lower than approximately 205 °C, the RAF mobilization promotes recrystallization, which becomes slower if RAF is absent at the amorphous/crystal interphase.
A confirmation that different reorganization mechanisms operate during heating is given in the literature also for isotactic polystyrene (iPS) [65]. FTIR measurements clearly revealed that recrystallization processes occur during heating in iPS samples crystallized at T c < 200 °C, whereas recrystallization during heating appears inhibited when isothermal crystallization is conducted at T c = 200 °C. Also in this case, the transition between two different melting/recrystallization mechanisms is observed after crystallization at a temperature that corresponds exactly to the transition between the triple and double melting behavior of iPS.
Conclusions
In the present study, the double and triple melting behavior that is observed after isothermal crystallization at low and high T c s, respectively, in PBT and iPS has been put into relation with the absence and presence of a vitrified rigid amorphous interphase coupled to original crystals. The discontinuity in the melting behavior has been rationalized by assuming that crystallizations at low temperatures involve partial vitrification of the amorphous chain portions coupled to the crystal lamellae. Conversely, at high T c s, crystallization takes place without or with a small fraction of amorphous chain segments subjected to geometrical constraints.
The complex melting behavior has been explained by supposing that a dual population of primary crystals (chain cluster aggregations and/or crystals with irregular adjacent reentry folding, and a high percentage of rigid amorphous fraction on the boundaries and crystals with a high percentage of regular adjacent reentry folding and reduced constraints at the amorphous/crystal interphase) develops during crystallization at low T c s, in a percentage that is a function of the crystallization temperature. These two distinct morphologies differently transform upon heating at low scanning rate, originating endotherms II and III. On the contrary, after crystallization at high T c s, a single reorganization route is followed by all the primary crystalline structures, characterized by the same morphology made of tightly chain-folded lamellae and absence of rigid amorphous fraction at the amorphous/crystal interphase.
References
Menczel Jd, Wunderlich B. Heat capacity hysteresis of semicrystalline macromolecular glasses. J Polym Sci Polym Lett. 1981;19:261–4.
Wunderlich B. Reversible crystallization and the rigid amorphous phase in semicrystalline macromolecules. Prog Polym Sci. 2003;28:383–450.
Di Lorenzo ML, Righetti MC. The three-phase structure of isotactic poly(1-butene). Polymer. 2008;49:1323–31.
Pieruccini M, Flores A, Nöchel U, Di Marco G, Stribeck N, Baltá-Calleja FJ. The role of the amorphous phase in the re-crystallization process of cold-crystallized poly(ethylene terephthalate). Eur Phys J E. 2008;27:365–73.
Kolesov I, Androsch R. The rigid amorphous fraction of cold-crystallized polyamide 6. Polymer. 2012;53:4770–7.
Martin S, Exposito MT, Vega JF, Martinez-Salazar J. Microstructure and properties of branched polyethylene: application of a three-phase structural model. J Appl Polym Sci. 2013;128:1871–8.
Nguyen TL, Bédoui F, Mazeran P-E, Guigon M. Mechanical investigation of confined amorphous phase in semicrystalline polymers: case of PET and PLA. Polym Eng Sci. 2015;55:397–405.
Lin J, Shenogin S, Nazarenko S. Oxygen solubility and specific volume of rigid amorphous fraction in semicrystalline poly(ethylene terephthalate). Polymer. 2002;43:4733–43.
Olson BG, Lin J, Nazarenko S, Jamieson AM. Positron annihilation lifetime spectroscopy of poly(ethylene terephthalate): contributions from rigid and mobile amorphous fractions. Macromolecules. 2003;36:7618–23.
Drieskens M, Peeters R, Mullen J, Franco D, Lemstra PJ, Hristova-Bogaerds DG. Structure versus properties relationship of poly(lactic acid). I. Effect of crystallinity on barrier properties. J Polym Sci Polym Phys. 2009;7:2247–58.
Delpouve N, Stoclet G, Saiter A, Dargent E, Marais S. Water barrier properties in biaxially drawn poly(lactic acid) films. J Phys Chem B. 2012;116:4615–25.
Guinault A, Sollogoub C, Ducruet V, Domenek S. Impact of crystallinity of poly(lactide) on helium and oxygen barrier properties. Eur Polym J. 2012;48:779–88.
Schick C, Wurm A, Mohammed A. Formation and disappearance of the rigid amorphous fraction in semicrystalline polymers revealed from frequency dependent heat capacity. Thermochim Acta. 2003;396:119–32.
Androsch R, Wunderlich B. The link between rigid amorphous fraction and crystal perfection in cold-crystallized poly(ethylene terephthalate). Polymer. 2005;46:12556–66.
Righetti MC, Tombari E, Angiuli M, Di Lorenzo ML. Enthalpy-based determination of crystalline, mobile amorphous and rigid amorphous fractions in semicrystalline polymers: poly(ethylene terephthalate). Thermochim Acta. 2007;462:15–24.
Chen H, Cebe P. Vitrification and devitrification of rigid amorphous fraction of PET during quasi-isothermal cooling and heating. Macromolecules. 2009;42:288–92.
Xu H, Ince S, Cebe P. Development of the crystallinity and rigid amorphous fraction in cold-crystallized isotactic polystyrene. J Polym Sci Polym Phys. 2003;41:3026–36.
Xu H, Cebe P. Heat capacity study of isotactic polystyrene: dual reversing crystal melting and relaxation of rigid amorphous fraction. Macromolecules. 2004;37:2797–806.
Righetti MC, Tombari E, Di Lorenzo ML. Crystalline, mobile amorphous and rigid amorphous fractions in isotactic polystyrene. Eur Polym J. 2008;44:2659–26676.
Righetti MC, Tombari E. Crystalline, mobile amorphous and rigid amorphous fractions in poly(l-lactic acid) by TMDSC. Thermochim Acta. 2011;522:118–27.
Di Lorenzo ML, Gazzano M, Righetti MC. The role of the rigid amorphous fraction on cold crystallization of poly(3-hydroxybutyrate). Macromolecules. 2012;45:5684–91.
Righetti MC, Tombari E, Di Lorenzo ML. The role of the crystallization temperature on the nanophase structure evolution of poly[(R)-3-hydroxybutyrate]. J Phys Chem B. 2013;117:12303–11.
Righetti MC, Laus M, Di Lorenzo ML. Temperature dependence of the rigid amorphous fraction in poly(ethylene terephthalate). Eur Polym J. 2014;58:60–8.
Bras AR, Malik P, Dionisio M, Mano JF. Influence of crystallinity in molecular motions of poly(l-lactic acid) investigated by dielectric relaxation spectroscopy. Macromolecules. 2008;41:6419–30.
Hong P-D, Chuang W-T, Yeh W-J, Lin T-L. Effect of rigid amorphous phase on glass transition behaviour of poly(trimethylene terephthalate). Polymer. 2002;43:6879–86.
Sanz A, Nogales A, Ezquerra TA, Lotti N, Munari A, Funari SS. Order and segmental mobility during polymer crystallization: poly(butylene isophthalate). Polymer. 2006;47:1281–90.
Henricks J, Boyum M, Zheng W. Crystallization kinetics and structure evolution of polylactic acid during melt and cold crystallization. J Therm Anal Calorim. 2015;120:1765–74.
Di Lorenzo ML, Righetti MC. Evolution of crystal and amorphous fractions of poly[(R)-3-hydroxybutyrate] upon storage. J Therm Anal Calorim. 2013;112:1439–46.
Di Lorenzo ML, Righetti MC, Cocca M, Wunderlich B. Coupling between crystal melting and rigid amorphous fraction mobilization in poly(ethylene terephthalate). Macromolecules. 2013;43:7689–94.
Pyda M, Nowak-Pyda E, Heeg J, Huth H, Minakov AA, Di Lorenzo ML, Schick C, Wunderlich B. Melting and crystallization of poly(butylene terephthalate) by temperature-modulated and superfast calorimetry. J Polym Sci Polym Phys. 2006;44:1364–77.
Pan P, Inoue Y. Polymorphism and isomorphism in biodegradable polyesters. Prog Polym Sci. 2009;34:605–40.
Müller AJ, Arnal ML. Thermal fractionation of polymers. Prog Polym Sci. 2005;30:559–603.
Righetti MC, Laus M. The biphasic behaviour of a thermotropic polymesomorphic polyester I. Time evolution of phase separation. Polymer. 2000;41:8355–62.
Righetti MC, Boggioni A, Laus M, Antonioli D, Sparnacci K, Boarino L. Thermal and mechanical properties of PES/PTFE composites and nanocomposites. J Appl Polym Sci. 2013;130:3624–33.
Capaccio G, Ward IM. Preparation of ultra-high modulus linear polyethylenes; effect of molecular weight and molecular weight distribution on drawing behaviour and mechanical properties. Polymer. 1974;15:233–8.
Zhou C, Clough SB. Multiple melting endotherms of poly(ethylene terephthalate). Polym Eng Sci. 1988;28:65–8.
Kong Y, Hay JN. Multiple melting behaviour of poly(ethyelene terephthalate). Polymer. 2003;44:623–33.
Minakov AA, Mordvintsev DA, Schick C. Melting and reorganization of poly(ethylene terephthalate) on fast heating (1000 K/s). Polymer. 2004;45:3755–63.
Nichols ME, Robertson RE. The multiple endotherms from poly(butylene terephthalate). J Polym Sci Polym Phys. 1992;30:755–68.
Righetti MC, Di Lorenzo ML, Angiuli M, Tombari E. Structural reorganization in poly(butylene terephthalate) during fusion. Macromolecules. 2004;37:9027–33.
Lemstra PJ, Kooistra T, Challa G. Melting behaviour of isotactic polystyrene. J Polym Sci Polym Phys. 1972;10:823–33.
Minakov AA, Mordvintsev DA, Tol R, Schick C. Melting and reorganization of the crystalline fraction and relaxation of the rigid amorphous fraction of isotactic polystyrene on fast heating (30,000 K/min). Thermochim Acta. 2006;442:25–30.
Pearce R, Marchessault RH. Multiple melting in blends of isotactic and atactic poly(β-hydroxybutyrate). Polymer. 1994;35:3990–7.
de Koning GJM, Scheeren AHC, Lemstra PJ, Peeters M, Reynaers H. Crystallization phenomena in bacterial poly[(R)-3-hydroxybutyrate]: 3. Toughening via texture changes. Polymer. 1994;35:4598–605.
Sawayanagi T, Tanaka T, Iwata T, Abe H, Doi Y, Ito K, Fujisawa T, Fujita M. Real-time synchrotron SAXS and WAXS studies on annealing behaviour of poly[(R)-3-hydroxybutyrate] single crystals. Macromolecules. 2006;39:2201–8.
Medellin-Rodriguez FJ, Phillips PJ, Lin JS, Campos R. The triple melting behavior of poly(ethylene terephthalate): molecular weight effects. J Polym Sci Polym Phys. 1997;35:1757–74.
Liu T, Petermann J. Multiple melting behavior in isothermally cold-crystallized isotactic polystyrene. Polymer. 2001;42:6453–61.
Sarge SM, Hemminger W, Gmelin E, Höhne GWH, Cammenga HK, Eysel W. Metrologically bases procedures for the temperature, heat and heat flow rate calibration of DSC. J Therm Anal. 1997;49:1125–34.
Mamum A, Okui N, Khan MAK. Crystallization kinetics of isotactic polystyrene from molten and glassy states. Can Chem Trans. 2013;1:267–76.
Wurm A, Merzlyakov M, Schick C. Reversible melting probed by temperature modulated dynamic mechanical and calorimetric measurements. Colloid Polym Sci. 1998;276:289–96.
Androsch R, Moon I, Kreitmeier S, Wunderlich B. Determination of heat capacity with a sawtooth–type, power compensated temperature modulated DSC. Thermochim Acta. 2000;357–358:267–78.
Pyda M, The advanced thermal analysis system (ATHAS) databank—polymer thermodynamics. In: Springer materials. Berlin: Springer; 2014. http://materials.bibliotecabuap.elogim.com/polymerthermodynamics.
Pyda M, Nowak-Pyda E, Mays J, Wunderlich B. Heat capacity of poly(butylene terephthalate). J Polym Sci Polym Phys. 2004;42:4401–11.
Di Lorenzo MC, Wunderlich B. Melting of polymers by non-isothermal, temperature-modulated calorimetry: analysis of various irreversible latent heat contributions to the reversing heat capacity. Thermochim Acta. 2003;405:255–68.
Righetti MC, Di Lorenzo ML, Tombari E, Angiuli M. The low-temperature endotherm in poly(ethylene terephthalate): partial melting and rigid amorphous fraction mobilization. J Phys Chem B. 2008;112:4233–41.
Righetti MC. Reversible melting in poly(butylene terephthalate). Thermochim Acta. 1999;330:131–5.
Wunderlich B. Methodology of interpreting thermal analysis of polymers. J Therm Anal Calorim. 2011;106:85–91.
Mathot VBF. Thermal characterization of states of matter. In: Mathot VBF, editor. Calorimetry and thermal analysis of polymers. Munich: Hanser Verlag; 1994. p. 105–67.
Righetti MC, Laus M, Di Lorenzo ML. Rigid amorphous fraction and melting behaviour of poly(ethylene terephthalate). Colloid Polym Sci. 2014;292:1365–74.
Flores A, Pieruccini M, Stribeck N, Funari SS, Bosch E, Baltá-Calleja FJ. Structure formation in poly(ethylene terephthalate) upon annealing as revealed by microindentation hardness and X-ray scattering. Polymer. 2005;46:9404–10.
Flores A, Pieruccini M, Nöchel U, Stribeck N, Baltá-Calleja FJ. Recrystallization studies on isotropic cold-crystallized PET: influence of heating rate. Polymer. 2008;49:965–73.
Fontaine F, Ledent J, Groeninckx G, Reynaers H. Morphological and melting behaviour of semicrystalline poly(ethylene terephthalate). 3. Quantification of crystal perfection and crystallinity. Polymer. 1982;23:185–91.
Jonas AM, Russell TP, Yoon DY. Time resolved SAXS studies of morphological changes in cold crystallized poly(ethylene terephthalate) during annealing and heating. Colloid Polym Sci. 1994;272:1344–51.
Kim HG, Robertson RE. Multiple melting endotherms in isothermally melt-crystallized poly(butylene terephthalate). J Polym Sci Polym Phys. 1998;36:1757–67.
Duan Y, Zhang Y, Shen D, Yan S. In situ FTIR studies on the cold-crystallization process and multiple melting behaviour of isotactic polystyrene. Macromolecules. 2003;36:4874–9.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Righetti, M.C., Di Lorenzo, M.L. Rigid amorphous fraction and multiple melting behavior in poly(butylene terephthalate) and isotactic polystyrene. J Therm Anal Calorim 126, 521–530 (2016). https://doi.org/10.1007/s10973-016-5553-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-016-5553-0