
Abstract
The thermal behavior of ammonium molybdates, i.e., (NH4)6Mo7O24·4H2O (1) and (NH4)2MoO4 (2), was studied in inert (N2) and oxidizing (air) atmospheres by TG/DTA-MS, XRD, FTIR and SEM. The thermal decomposition sequence of 2 had similarities to 1; however, there were significant differences as well. When both of them were annealed, NH3 and H2O were released parallel, and in air the as-evolved NH3 was burnt partially into NO and N2O. In both atmospheres, while 1 decomposed in four steps, the thermal decomposition of 2 involved 5 steps. In the case of 1, the intermediate products were (NH4)8Mo10O34, (NH4)2Mo4O13 and h-MoO3. In contrast, the decomposition intermediates of 2 were (NH4)2Mo3O10, (NH4)2Mo2O7, (NH4)2Mo4O13 and h-MoO3. By both 1 and 2, the final product was dominated by o-MoO3, accompanied with small amount of Mo4O11 in N2, which was absent in air. Most decomposition steps were endothermic, except for the last step around 400 °C, where crystallization from the residual amorphous phase had an exothermic heat effect. In addition, the combustion of NH3 also changed the DTA curve into exothermic in some cases. The morphology of the final products was characterized by 1–5 μm sheet-like particles, except for annealing 2 in N2, when 0.5- to 1-μm-thick and 5- to 10-μm-long needle-shaped particles were detected.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Ammonium molybdate tetrahydrate or ammonium heptamolybdate, i.e., (NH4)6Mo7O24·4H2O (1), is widely used for the preparation of molybdenum oxides, reduced molybdenum oxides, mixed molybdenum oxides or supported molybdenum oxide catalysts, and it can be also a precursor for molybdenum carbide or molybdenum metal. MoOx catalysts have applications in various catalytic reactions, e.g., partial oxidation for light alkenes, hydrotreating, hydrodenitrogenation or hydrodesulfurization [1–5]. Molybdenum carbide is also applied as a catalyst for, e.g., hydrogenation reactions [6, 7]. Metallic molybdenum offers excellent mechanical, thermal and electrical properties and good corrosion resistance which makes it attractive for electronics, metallurgical, aerospace and electrical industries [8].
The structure of (NH4)6Mo7O24·4H2O is composed of monomers of seven distorted, edge-sharing, molybdenum-oxygen octahedra. The thermal behavior of (NH4)6Mo7O24·4H2O has been studied in a great number of papers [9–38]. The role of decomposition conditions (e.g., atmosphere, heating rate, the presence of dopants, and influence of possible support materials) has been investigated in detail. The reason for this increased interest is that the various MoOx materials, molybdenum carbide and molybdenum metal can be easily obtained by the thermal treatment of (NH4)6Mo7O24·4H2O, and their application properties depend strongly on the structure, composition and morphology of the products, which are then determined by the circumstances of the annealing of the precursor [39].
However, there is another ammonium molybdate, i.e., (NH4)2MoO4 (2), called ammonium orthomolybdate, which has a much simpler structure and composition [40]. It is mostly used as a corrosion inhibitor and can be also an intermediate to gain Mo from its ores [40]. Interestingly, the thermal behavior of (NH4)2MoO4 has not been studied in detail yet, to the best of our knowledge. In order to use it also as a possible precursor for MoOx compounds, it is important to obtain information about its thermal properties.
Hence, in this study, our aim was to explore the thermal decomposition of (NH4)2MoO4 and also study the thermal properties of (NH4)6Mo7O24·4H2O for comparison. Thus, the annealing of the two materials was studied in both inert (N2) and oxidizing (air) atmospheres. The thermal events were followed by simultaneous thermogravimetry and differential thermal analysis (TG/DTA) and also by evolved gas analysis performed by a mass spectrometer (EGA-MS) coupled on-line to the TG/DTA device. The changes in morphology, structure and bonds were investigated by scanning electron microscopy (SEM), X-ray powder diffraction (XRD) and infrared spectroscopy (FTIR), respectively.
Experimental
The (NH4)6Mo7O24·4H2O (1) and (NH4)2MoO4 (2) samples were obtained from Sigma-Aldrich, and they were used as received.
SEM images were recorded by a JEOL JSM-5500LV scanning electron microscope.
Powder XRD patterns were measured on a PANalytical X’pert Pro MPD X-ray diffractometer using Cu Kα radiation.
FTIR spectra were obtained by an Excalibur Series FTS 3000 (Biorad) FTIR spectrophotometer in the range of 400–4000 cm−1 in KBr pellets.
The thermal decomposition of the samples was studied by a TG/DTA-MS apparatus, which consisted of an STD 2960 Simultaneous TGA/DTA (TA Instruments Inc.) thermal analyzer and a Thermostar GSD 200 (Balzers Instruments) quadrupole mass spectrometer. On-line coupling between the two parts was provided through a heated (T = 200 °C) 1 m 100 % methyl deactivated fused silica capillary tube with inner diameter of 0.15 mm. A mass range between m/z = 1–200 was monitored by scan mode. During the measurements, an open platinum crucible, a heating rate of 10 °C min−1, sample sizes of 5–6 mg and flowing air (130 mL min−1) were used.
Results and discussion
Characterization of 1
According to SEM images (Fig. 1a), sample 1 consisted of irregularly shaped 10–50 μm aggregated particles. The sample was made up only by (NH4)6Mo7O24·4H2O (ICDD 27-1013), and there were no impurities detected in the X-ray powder diffractogram (Fig. 2). In the FTIR spectrum (Fig. 3), the bands below 1000 cm−1 were assigned to various Mo–O lattice vibrations, characteristic to polymolybdates [41]. The N–H and O–H deformation bands of ammonium ions and water appeared at 1400 and 1630 cm−1, respectively, while the N–H and O–H stretching vibrations were detected at 3135 and 3570 cm−1 [42, 43].

SEM images of a 1, b 1 annealed in N2 at 700 °C, c 1 annealed in air at 700 °C

XRD patterns of 1 and its decomposition products in N2

FTIR spectra of 1 and its decomposition products in N2
Thermal decomposition of 1 in N2
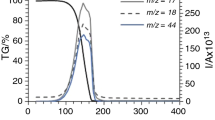
The first decomposition step was observed between 25 and 180 °C, and it was accompanied by an endothermic heat effect (Fig. 4). NH3 and H2O were released parallel. Due to their evolution, the intensity of the N–H and O–H bands in the FTIR spectrum decreased to some extent. The initial crystal structure changed completely, and a new polymolybdate phase, i.e., (NH4)8Mo10O34 (ICDD 37-0381), appeared (93.3 and 93.2 % theoretical and measured remaining mass), as evidenced by XRD and FTIR.

TG/DTA and EGA-MS curves of 1 in N2
In the second decomposition step (180–270 °C) in an endothermic reaction, again NH3 and H2O evolved, and the (NH4)8Mo10O34 phase transformed into (NH4)2Mo4O13 (ICDD 11-4665) (88.9 and 88.7 % theoretical and measured remaining mass). The N–H and O–H FTIR peaks reduced in intensity considerably, and the Mo–O lattice vibration region was also modified.
In the third, endothermic decomposition step (270–370 °C), the parallel evolution of NH3 and H2O continued, and the O–H and N–H bands in the infrared spectrum disappeared almost completely. The crystal structure changed, and a mixture of two phases was present. Traces of hexagonal molybdenum oxide, i.e., h-MoO3 (ICDD 21-0569) were observed, but besides this phase already orthorhombic molybdenum oxide, i.e., o-MoO3 (ICDD 12-8070), also appeared, which was the main component at this temperature.
In the last step (370–400 °C) in a slightly exothermic reaction, the amount of o-MoO3 increased (81.5 and 81.2 % theoretical and measured remaining mass). The h-MoO3 phase disappeared, and a partially reduced molybdenum oxide, i.e., Mo4O11 (ICDD 89-8980), formed, which was responsible for the lower measured remaining mass than the theoretical value for MoO3. FTIR also revealed the formation of molybdenum oxide [44].
At the end of the annealing, the morphology changed significantly compared to the starting material (Fig. 1b). At 700 °C, the product was built up by 1–5 μm particles and sheets.
Thermal decomposition of 1 in air
The first two steps of the thermal decomposition of 1 in air were similar to those in N2. From the starting material in the first endothermic step (25–180 °C), NH3 and H2O were released (Fig. 5) and (NH4)8Mo10O34 formed (Fig. 6). Then, between 180 and 270 °C, this phase transformed into (NH4)2Mo4O13 in an endothermic reaction, accompanied by the release of NH3 and H2O. In the FTIR spectra (Fig. 7), the changes were also very similar to those in air; the most remarkable was the decrease in the intensity of the N–H and O–H vibrations, and as the crystal structure changed, the lattice vibration region below 1000 cm−1 was modified. The only significant difference was that in air in the first decomposition step a small amount of the as-released NH3 was oxidized thermally to NO. Due to the elevated temperature, in the second decomposition step, a larger portion of the as-evolved NH3 was burnt, and besides NO also N2O was detected as combustion product. Although the oxidation of NH3 is an exothermic process [45–47], still the endothermic heat effect of the decomposition of the solid phase was dominant on the DTA curve; thus, here again an intense endothermic DTA peak was observed at 234 °C.

TG/DTA and EGA-MS curves of 1 in air

XRD patterns of 1 and its decomposition products in air

FTIR spectra of 1 and its decomposition products in air
It must be mentioned that N2O and NO formation might be also the consequence of NH4NO3 intermediate formation and decomposition to some extent. Such phenomenon was observed in case of ammonium salts of other oxometallates (e.g., permanganates, vanadates), when the metal oxide-type intermediates could catalyze the oxidation reaction [48–52].
In contrast to inert atmosphere, in air in the third step (270–350 °C), the (NH4)2Mo4O13 phase transformed only into o-MoO3. The absence of h-MoO3 at the end of the third decomposition step is explained by annealing in oxidizing atmosphere. Due to annealing in air, besides NH3 and H2O, also NO and N2O were detected, similar to the second decomposition step.
In the fourth step, similar to N2 atmosphere, a small exothermic heat effect was observed. However, here in the final solid product, only o-MoO3 was observed without the partially reduced Mo4O11, in contrast to what was observed in inert atmosphere; thus, here the measured remaining mass was equal to the theoretical 81.5 % value. Before and after the exothermic reaction, the solid phase contained o-MoO3, and the only difference was that the sample became more crystalline at the end of the fourth step. Hence, the exothermic heat effect is explained by the further crystallization of o-MoO3 from the residual amorphous part of the sample at 350 °C.
The morphology of the final product was characterized again by 1–5 μm particles; however, here their size was even smaller than in N2, and most of them were sheet like.
Characterization of 2
Sample 2 was built up by 1–10 μm particles, which were aggregated into much larger blocks (Fig. 8a). Thus, the particles size of sample 2 was much smaller than that of 1. The sample consisted of only (NH4)2MoO4 (ICDD 12-2248) with no impurities. The FTIR spectrum was similar to 1; however, in 2, the O–H deformation vibration band (1630 cm−1) was more intense compared to the N–H deformation peak (1400 cm−1). The Mo–O vibration region below 1000 cm−1 was also somewhat different, corresponding to the difference in the crystal structures of 1 and 2 (Figs. 9, 10).

SEM images of a 2, b 2 annealed in N2 at 700 °C, c 2 annealed in air at 700 °C

XRD patterns of 2 and its decomposition products in N2

FTIR spectra of 2 and its decomposition products in N2
Thermal decomposition of 2 in N2
Though the structure and composition of 2 were quite simple, nevertheless, its thermal decomposition was quite complex, which is in contrast to, e.g., (NH4)2WS2, which has similarly simple structure and composition and its thermal behavior is not complicated and involves only few decomposition steps and intermediates [53].
The mass loss (17.6 %) in the first decomposition (25–200 °C) step of 2 in N2 (Fig. 11) was much larger than in the case of 1. Similarly, NH3 and H2O were released parallel in an endothermic reaction, and in the FTIR spectrum, the intensity of the N–H and O–H peaks decreased to some extent. The intermediate sample after the first decomposition step consisted of different phases, compared to 1, i.e., (NH4)2Mo3O10 (ICDD 09-9568), and a small amount of (NH4)2Mo2O7 (ICDD 12-7094) were detected at 200 °C in the case of annealing 2 in N2.

TG/DTA and EGA-MS curves of 2 in N2
In the second decomposition step (200–230 °C), accompanied with a small evolution of NH3 and H2O and 0.7 % mass loss, in an endothermic reaction, the (NH4)2Mo2O7 content of the sample at 200 °C transformed into (NH4)2Mo3O10. Thus, the sample at 230 °C contained only (NH4)2Mo3O10 (82.3 and 81.7 % theoretical and measured remaining mass).
The third decomposition step (230–280 °C) was also characterized by a small mass loss (2.4 %). Here again NH3 and H2O were detected as gaseous decomposition products. The sample at 280 °C contained only (NH4)2Mo4O13 (80.1 and 79.4 % theoretical and measured remaining mass), which was also the only constituent of the intermediate sample at 270 °C during the thermal decomposition of 1 in N2. Therefore, from this stage, the thermal behavior of 2 has become similar to 1.
Accordingly, sample 2 almost completely lost its NH +4 and H2O contents at 370 °C in an endothermic reaction, and hence, the N–H and O–H deformation and stretching FTIR peaks basically disappeared. As the main constituent of the intermediate sample at 370 °C, o-MoO3 was detected; and in addition, traces of h-MoO3 were also present.
Then, in the final decomposition step (370–420 °C), the h-MoO3 disappeared, the o-MoO3 became more crystalline accompanied by an exothermic heat effect (exothermic DTA peak at 400 °C) and also the Mo4O11 phase was detected to some extent, just as in the case of 1. Accordingly, the remaining mass was a bit lower (73.3 %) than what expected for pure MoO3 (73.5 %).
In contrast to 1, the morphology of the final decomposition product of 2 was dominated by needle-like particles, which were 0.5–1 μm thick and 5–10 μm long (Fig. 8b).
Thermal decomposition of 2 in air
The thermal decomposition of 2 in air was a combination of the thermal behavior of 2 in N2 and the thermal decomposition of 1 in air.
In the first decomposition step (25–205 °C), NH3 and H2O were released parallel and also small amount of NO was detected (Fig. 12). The mass loss (15.8 %) was similar as in N2, and the formed crystalline phases were also similar, i.e., (NH4)2Mo3O10 and (NH4)2Mo2O7 (Fig. 13). The intensity of the N–H and O–H stretching and deformation modes decreased to some extent, and there were also changes in the Mo–O vibration region below 1000 cm−1, according to the change in the crystalline structure (Fig. 14).

TG/DTA and EGA-MS curves of 2 in air

XRD patterns of 2 and its decomposition products in air

FTIR spectra of 2 and its decomposition products in air
In the second (205–230 °C) and third decomposition (230–270 °C) steps, NH3 and H2O evolved (endothermic DTA peaks at 214 and 242 °C, respectively), and at 270 °C the solid sample was composed of mostly (NH4)2Mo4O13.
In the fourth decomposition step (270–370 °C), the sample transformed into o-MoO3. Here the combustion of the as-released NH3 was so intensive that the DTA peak started as endothermic (endothermic DTA peak at 317 °C) due to the decomposition of the solid materials, but then it changed into exothermic at the second part of the decomposition step (exothermic DTA peak at 343 °C). The XRD pattern of the intermediate obtained at 325 °C showed traces of h-MoO3, but by the end of this step these peaks disappeared.
In the last decomposition step, the residual amorphous part of the sample transformed into o-MoO3 in an exothermic reaction (small exothermic DTA peak at 420 °C), and hence, the XRD peaks of this phase became stronger and narrower, and FTIR also showed the characteristic bands of the o-MoO3 phase.
Unlike annealing of 2 in N2, the morphology of the final product of 2 in air was built up again by sheet-like particle of 1–5 μm (Fig. 8c), similar to when 1 was annealed in N2 and air.
Conclusions
The thermal decomposition of (NH4)6Mo7O24·4H2O (1) and (NH4)2MoO4 (2) was studied extensively in inert (N2) and oxidizing (air) atmospheres by TG/DTA-MS, XRD, FTIR and SEM.
1 decomposed in four steps in N2, where NH3 and H2O were released parallel. The intermediate products were (NH4)8Mo10O34, (NH4)2Mo4O13 and h-MoO3, while the final sample was dominated by o-MoO3, accompanied with a small amount of the partially reduced Mo4O11 phase. In air, the as-released NH3 was oxidized thermally into NO and N2O. Due to the oxidizing atmosphere, the only final product was o-MoO3. The hexagonal molybdenum oxide phase (h-MoO3) and the partially reduced molybdenum oxide (Mo4O11) in air were not observed.
The thermal decomposition sequence of 2 had similarities to 1; however, there were significant differences as well. In addition, though the structure and composition of 2 were quite simple, its thermal decomposition was complex. In N2, the decomposition intermediates were (NH4)2Mo3O10 and (NH4)2Mo2O7 in the first two steps in contrast to 1, where only (NH4)8Mo10O34 was observed as a decomposition intermediate in this region. Then, at 270–280 °C, (NH4)2Mo4O13 was a common intermediate for both 1 and 2. The final product was o-MoO3 together with a small amount of Mo4O11, similar to 1. The thermal decomposition of 2 in air was a combination of the thermal behavior of 2 in N2 and the thermal decomposition of 1 in air. The decomposition intermediates were the same as for 2 in N2, i.e., (NH4)2Mo3O10, (NH4)2Mo2O7, (NH4)2Mo4O13 and h-MoO3, and the final product was highly crystalline o-MoO3 without the presence of Mo4O11. Similar to the decomposition of 1 in air, the as-evolved NH3 was burnt partially into NO and N2O.
In the case of both 1 and 2, most decomposition steps were endothermic. An exception was the last step for both phases (around 400 °C), where the increase in the crystallinity of the samples through the formation of o-MoO3 from the residual amorphous phase at the end of the previous step had exothermic heat effect. In addition, the combustion of NH3 was so intensive that, when 2 was annealed in air, after 330 °C the DTA curve changed from endothermic to exothermic.
While 1 consisted of irregularly shaped 10–50 μm aggregated particles, 2 was built up by 1–10 μm particles. Nevertheless, after annealing 1 in air and N2 and heating 2 in air, the final products were composed of 1–5 μm particles and sheets. In contrast, when 2 was annealed in N2 at 700 °C, the sample was dominated by 0.5- to 1-μm-thick and 5- to 10-μm-long needle-like particles.
References
Liu H, Liu C, Yin C, Chai Y, Li Y, Liu D, Liu B, Li X, Wang Y, Li X. Preparation of highly active unsupported nickel–zinc–molybdenum catalysts for the hydrodesulfurization of dibenzothiophene. Appl Catal B. 2015;174–175:264–76.
Adamiak J. Controlled nitration of anisole over HNO3/PO4/MoO3/SiO2/solvent systems. J Mol Catal A. 2015;407:81–6.
Ressler T, Walter A, Huang ZD, Bensch W. Structure and properties of a supported MoO3–SBA-15 catalyst for selective oxidation of propene. J Catal. 2008;254:170–9.
Grasselli RK. Advances and future trends in selective oxidation and ammoxidation catalysis. Catal Today. 1999;49:141–53.
Haber J, Lalik E. Catalytic properties of MoO3 revisited. Catal Today. 1997;33:119–37.
Zaman S, Smith K. A review of molybdenum catalysts for synthesis gas conversion to alcohols: catalysts, mechanisms and kinetics. Catal Rev Sci Eng. 2012;54:41–132.
Kim HG, Lee KH, Lee JS. Carbon monoxide hydrogenation over molybdenum carbide catalysts. Res Chem Intermed. 2000;26:427–43.
Saji VS, Lopatin SI, editors. Molybdenum and its compounds: applications, electrochemical properties and geological implications. New York: Nova Science Publishers; 2014.
Hegedűs AJ, Sasvári K, Neugebauer J. Thermo- und röntgenanalytischer Beitrag zur Reduktion des Molybdäntrioxyds und zur Oxydation bzw. Nitrierung des Molybdäns. Z Anorg Allg. Chemie. 1957;293:56–83.
Onchi M, Ma E. An application of the omegatron mass spectrometer to thermal decomposition studies. J Phys Chem. 1963;67:2240–1.
Ma E. Thermal decomposition of ammonium polymolybdates I. Bull Chem Soc Jpn. 1964;37:171–5.
Ma E. Thermal decomposition of ammonium polymolybdates II. Bull Chem Soc Jpn. 1964;37:648–53.
Schwing-Weill MJ. Bull Soc Chim Fr. 1967;10:3795–8.
Kiss AB, Gadó P, Asztalos I, Hegedűs AJ. Acta Chim Acad Sci Hung. 1970;66:235–49.
Louisy A, Dunoyer JM. Thermal decomposition of ammonium paramolybdate. Bull Soc Chim Fr. 1970;67:1390–4.
Bhatnagar IK, Chakrabarty DK, Biswas AB. Thermal decomposition of ammonium vanadate, ammonium molybdate, and ammonium tungstate. Indian J Chem. 1972;10:1025–8.
Hanafi ZM, Khilla MA, Askar MH. The thermal decomposition of ammonium heptamolybdate. Thermochim Acta. 1981;45:221–32.
Isa K, Ishimura H. Thermal decomposition studies of ammonium heptamolybdate(6-) tetrahydrate by means of high-temperature oscillating x-ray diffraction with a rotating anode type large capacity generator. Bull Chem Soc Jpn. 1981;54:3628–34.
Topic M, Mogus-Milankovic A. A multiple thermal-analysis of ammonium heptamolybdate tetrahydrate. Chroatica Chem Acta. 1984;57:75–83.
Sharma IB, Batra S. Characterization and thermal investigations of ammonium heptamolybdate. J Therm Anal. 1987;34:1273–81.
Yong WJ. The GC study of the thermal decomposition of ammonium paramolybdate tetrahydrate in a hydrogen atmosphere. Thermochim Acta. 1990;158:183–6.
Shashkin DP, Shiryaev PA, Kutyrev MY, Krylov OV. Peculiarities of the effect of vanadium ions on molybdena development under prereaction conditions. Kin Catal. 1993;34:302–6.
Halawy SA, Mohamed MA. Characterization of unsupported molybdenum oxide—cobalt oxide catalysts. J Chem Tech Biotechnol. 1993;58:237–45.
Said AA, Halawy SA. Effects of alkali metal ions on the thermal decomposition of ammonium heptamolybdate tetrahydrate. J Therm Anal. 1994;41:1075–90.
Said AA. Mutual influences between ammonium heptamolybdate and g-alumina during their thermal treatments. Thermochim Acta. 1994;236:93–104.
Cabello CI, Botto IL, Thomas HJ. Reducibility and thermal behavior of some Anderson phases. Thermochim Acta. 1994;232:183–93.
Bi M, Li H, Pan WP, Lloyd WG, Davis BH. Thermal studies of (NH4)2Cr2O7, (NH4)2WO4 and (NH4)6Mo7O24·4H2O deposited on ZrO2. Thermochima Acta. 1996;284:153–60.
Valmalette JC, Houriet R, Hofmann H, Gavarri JR. Formation of N2O during the thermal decomposition of ammonium salts (NH4)aMxOy (M = V, Cr, Mo and W). Eur J Solid State Inorg Chem. 1997;34:317–29.
Li JL, Inui T. Characterization of precursors of methanol synthesis catalysts, copper/zinc/aluminum oxides, precipitated at different pHs and temperatures. Appl Catal A. 1996;137:105–17.
Zhoulan Y, Xinhai L, Qiyuan C. Study on the kinetics of the thermal decompositions of ammonium molybdates. Thermochim Acta. 2000;352–353:107–10.
Thomazeau C, Martin V, Afanasiev P. Effect of support on the thermal decomposition of (NH4)6Mo7O24·4H2O in the inert gas atmosphere. Appl Cat A. 2000;199:61–72.
Shaheen WM, Selim MM. Thermal decompositions of pure and mixed manganese carbonate and ammonium molybdate tetrahydrate. J Therm Anal Calorim. 2000;59:961–70.
Murugan R, Chang H. Thermo-Raman investigations on thermal decomposition of (NH4)6Mo7O24·4H2O. J Chem Soc, Dalton Trans. 2001;20:3125–32.
Shaheen WM. Thermal behaviour of pure and binary basic nickel carbonate and ammonium molybdate systems. Mater Lett. 2002;52:272–82.
Wienold J, Jentoft RE, Ressler T. Structural investigation of the thermal decomposition of ammonium heptamolybdate by in situ XAFS and XRD. Eur J Inorg Chem. 2003; 1058–1071.
Radwan NRE, Mokhtar M, El-Shobaky GA. Thermal behaviour of ammonium molybdate-basic magnesium carbonate system doped with lithium nitrate. J Therm Anal Calorim. 2003;71:977–86.
Manukyan K, Davtyan D, Bossert J, Kharatyan S. Direct reduction of ammonium molybdate to elemental molybdenum by combustion reaction. Chem Eng J. 2011;168:925–30.
Biedunkiewicz A, Krawczyk M, Gabriel-Polrolniczak U, Figiel P. Analysis of (NH4)6Mo7O24·4H2O thermal decomposition in argon. J Therm Anal Calorim. 2014;116:715–26.
Gaigneaux EM, Genet MJ, Ruiz P, Delmon B. Catalytic behavior of molybdenum suboxides in the selective oxidation of isobutene to methacrolein. J Phys Chem B. 2000;104:5724–37.
Sebenik RF, Burkin AR, Dorfler RR, Laferty JM, Leichtfried G, Meyer-Grünow H, Mitchell PCH, Vukasovich MS, Church DA, Van Riper GG, Gilliland JC, Thielke SA. Molybdenum and Molybdenum Compounds. In Ullmann’s Encyclopedia of Chemical Technology. Weinheim: Wiley-VCH; 2005.
Alizadeh S, Hassanzadeh-Tabrizi SA. Ceram Int. 2015;41:10839–43.
Szilágyi IM, Madarász J, Hange F, Pokol G. Partial thermal reduction of ammonium paratungstate tetrahydrate. J Therm Anal Calorim. 2007;88:139–44.
Hunyadi D, Sajó I, Szilágyi IM. Structure and thermal decomposition of ammonium metatungstate. J Therm Anal Calorim. 2014;116:329–37.
Chianga TH, Hob PY, Chiub SY, Chaob AC. Synthesis, characterization and photocatalytic activity of α–MoO3 particles utilizing different polyol monomers under visible light irradiation. J Alloy Comp. 2015;651:106–13.
Szilágyi IM, Santala E, Heikkilä M, Kemell M, Nikitin T, Khriachtchev L, Räsänen M, Ritala M, Leskelä M. Thermal study on electrospun polyvinylpyrrolidone/ammonium metatungstate nanofibers: optimising the annealing conditions for obtaining WO3 nanofibers. J Therm Anal Calorim. 2011;105:73–81.
Madarász J, Szilágyi IM, Hange F, Pokol G. Comparative evolved gas analyses (TG-FTIR, TG/DTA-MS) and solid state (FTIR, XRD) studies on thermal decomposition of ammonium paratungstate tetrahydrate (APT) in air. J Anal Appl Pyrol. 2004;72:197–201.
Szilágyi IM, Madarász J, Pokol G, Király P, Tárkányi G, Saukko S, Mizsei J, Tóth AL, Szabó A, Varga-Josepovits K. Stability and controlled composition of hexagonal WO3. Chem Mater. 2008;20:4116–25.
Range KJ, Zintl R. The thermal decomposition of ammonium metavanadate(V) in open and closed systems. Z Naturforsch. 1988;43:309–17.
Kótai L, Sajó IE, Jakab E, Keresztury G, Németh C, Gács I, Menyhárd A, Kristóf J, Hajba L, Petrusevski VM, Ivanovski V, Timpu D, Sharma PK. Studies on the chemistry of [Cd(NH3)4](MnO4)2. A low temperature synthesis route of the CdMn2O4+x Type NOx and CH3SH sensor precursors. Z Anorg Allg Chem. 2012;638:177–86.
Kótai L, Gács I, Sajó IE, Sharma PK, Banerji KK. Beliefs and facts in permanganate chemistry – an overview on the synthesis and the reactivity of simple and complex permanganates. Trend Inorg Chem. 2009;11:25–104.
Kótai L, Baneri KK, Sajo IE, Kristóf J, Sreedhar B, Holly S, Keresztury G, Rockenbauer A. An unprecedented-type intramolecular redox reaction of Solid Tetraamminecopper(2+) Bis(permanganate) ([Cu(NH3)4](MnO4)2): a low-temperature synthesis of copper dimanganese tetraoxide-type (CuMn2O4) nanocrystalline catalyst precursors. Helvet Chim Acta. 2002;85:2316–27.
Sajo IE, Kótai L, Keresztury G, Gács I, Pokol G, Kristóf J, Soptrayanov B, Petrusevski VM, Timpu D, Sharma PK. Studies on the chemistry of tetraamminezinc(II) Dipermanganate ([Zn(NH3)4](MnO4)2): low-temperature synthesis of the manganese zinc oxide (ZnMn2O4) catalyst precursor. Helvet Chim Acta. 2008;91:1646–58.
Hunyadi D, Ramos ALVM, Szilágyi IM. Thermal decomposition of ammonium tetrathiotungstate. J Therm Anal Calorim. 2015;120:209–2015.
Acknowledgements
I. M. Szilágyi thanks for a János Bolyai Research Fellowship of the Hungarian Academy of Sciences. An OTKA-PD-109129 grant is acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kovács, T.N., Hunyadi, D., de Lucena, A.L.A. et al. Thermal decomposition of ammonium molybdates. J Therm Anal Calorim 124, 1013–1021 (2016). https://doi.org/10.1007/s10973-015-5201-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-015-5201-0