
Abstract
This study deals with the effects of Ru–Bi partial substitutions on the thermal, structural and mechanical properties of Bi1.8−x Ru x Pb0.2Sr2CaCu2O10+δ (x = 0.0, 0.025, 0.050, 0.075) system, produced by using glass ceramics method. The effects of Ru–Bi partial substitutions on glass transition, nucleation and crystallization temperature were analyzed via differential thermal analyzer (DTA). Besides, microstructure and micromechanical properties of Ru–Bi partially substituted BSCCO glass ceramics were also investigated by X-ray diffraction (XRD), scanning electron microscopy (SEM) and Vickers microhardness measurements. From the DTA results, nucleation kinetics were studied by using Ozawa, Augis–Bennett, Takhor and Kissinger equations. In addition to this, activation energies and Avrami parameters were also calculated. According to the results of the thermogravimetric analyses, the amount of oxidation was seen to be increased with increasing Ru concentration. Moreover, the lattice parameters, volume fractions and surface morphologies of our newly produced glass ceramics were investigated by XRD and SEM measurements, respectively. RISE behavior obtained from the microhardness measurements for all samples. Microhardness, elastic modulus, yield strength and fracture toughness values decreased with increasing Ru doping.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Glass ceramics are polycrystalline materials, which are produced by the crystallization of glass. They have higher durability, higher impact resistance and lower thermal expansion coefficient than glass [1]. Ceramic materials are more resistant to corrosion and chemical effects at higher temperatures than metals. They may also have lower density values, depending on the type of their application. Nowadays, it has been pointed out that glass ceramic materials have many high technology applications in comparison with metals due to their superior corrosion, abrasion and impact resistance and toughness properties. Their specific area applications can be outlined as biomedical materials, superconducting materials, vacuum technology, bioactive glasses, high-dielectric-constant materials and some base plates used in electronics [2–6].
Hardness test is a generally used technique for the investigation of the mechanical properties of materials. The principle of indentation hardness testing is based on the application of load to the indenter and forming an indentation on the surface of the material. The hardness is calculated by using the sizes of the formed indentations. Indentation hardness test is accepted as a nondestructive test, since it produces only a small indention on the surface of a material under investigation.
Hardness is related to the structure and composition of materials; therefore, it is a popular method for the characterization of materials [7]. Three properties of materials are effective in indentation which are (1) the crystal structure of the grains, (2) grain orientation and (3) porosity among grains [8]. Since the hardness is a measure of porosity, the smaller value of hardness is obtained where porosity is high [9]. Moreover, validation of the hardness results obtained by XRD and SEM measurements must be followed also.
In this study, the effect of Bi-site Ru substitution on thermal, structural and mechanical properties of bulk Bi-2212 superconducting samples was investigated. Crystallization kinetic parameters for this type of Ru-doped glass ceramics were investigated first time, in this study. Small atomic radius of Ru, compared with Bi and Pb, may affect the mechanical, structural and thermal properties of samples. For this reason, the characterization of the sample is crucible for the application in the technology. Nowadays, usage of the Bi-based ceramic materials for producing cables shows the importance of mechanical characterization. In this respect, characterization of Ru-doped Bi-based glass ceramic structure will complete an important lack in the literature.
The aim of this study was to investigate the effect of Ru–Bi partial substitutions on the thermal, microstructural and micromechanical properties of Bi1.8−x Ru x Pb0.2Sr2CaCu2O10+δ glass ceramics by means of the differential thermal analyzer (DTA) which determines the thermal properties, Vickers microhardness measurements for mechanical properties, XRD measurements for the phase composition and lattice parameters and finally the scanning electron microscopy (SEM) for microstructural examination of the samples.
Experimental
Glass synthesis
Required amounts of high-purity Bi2O3, PbO, SrCO3, CaCO3, RuO2 and CuO powders were mixed in an agate mortar about 4 h to obtain the Bi1.8−xRuxPb0.2Sr2CaCu2O10+δ glass ceramic samples with x = 0.0, 0.025, 0.050, 0.075. The resulting mixture was placed into an alumina crucible and heated up to 1150 °C for 1.5 h with a heating rate of 12 °C min−1 in a programmable furnace under ordinary atmospheric conditions. The molten mixture was poured in between two cooled copper plates. Finally, 0.6–0.8 mm of thick black glasses were obtained. After this process, glass ceramic samples obtained by sintering the glass samples at 840 °C under an oxygen flow for 120 h in a furnace.
Characterization studies
The differential thermal analyses and thermogravimetric measurements of the glass ceramic samples (approximately 10 mg) were carried out by the DTA, model SII 7200 Exstar DTA/TG. Measurements were realized at four different heating rates under oxygen gas condition. The measurement errors were ±0.5 μV in DTA and ±0.0005 % in thermogravimetric measurements at the crystallization kinetic studies, respectively. Moreover, the phase composition of the samples was characterized by using a Bruker D8 Advance model X-ray diffractometer with CuKα radiation (λ = 1.5418 Å) in the range 2θ = 3°–90° at a scan speed of 3° min−1 and a step increment of 0.02° at room temperature. Phase purity and lattice parameters of these XRD patterns were determined via Bruker EVA 14.0.0.0 program. The accuracy in determining the lattice parameters (a, b and c) was found to be ±0.0001 Å. In addition, the grain sizes, separation distances between these grains, surface morphology and microstructure of the samples were measured and imaged by field emission gun (FEG) type, JEOL 7000F SEM. Moreover, microhardness measurements of the samples produced were taken by using a digital microhardness tester (Shimadzu HVM-2) at room temperature. The applied load was within the range of 0.245–2.940 N for a peak-load time of 10 s, and the diagonals of indentation were measured with an accuracy of ±0.1 μm. The Vickers microhardness values were determined as an average of 10 readings gathered from different locations of the sample surface. For simplicity, the abbreviations Ru0, Ru025, Ru050 and Ru075 will be used for the samples x = 0.0, 0.025, 0.050 and 0.075, respectively, in rest of the following text.
Theoretical background
In general, activation energy is the most important crystallization kinetic parameter almost for all amorphous materials and it is defined as a measure of the resistance of a material against the crystallization. Matsushita and Sakka [10] proved that it is important to know and take into account the growth mechanism of the glass. Otherwise, it is not possible to reach a significant value.
Kinetic studies are performed by DTA which can analyze with non-isothermal technique. Numerical data, belonging to crystallization and nucleation, can also be obtained with this method. DTA method is based on the ratio of temperature difference of the sample and reference material to temperature of the reference material [11–13]. Crystallization peaks, obtained as a function of heating rate, are displayed as sequent peaks with increasing heating rate. Crystallization ratio must be calculated separately for each analysis at different heating rates [14, 15]. Figure 1 shows how a typical crystallization peak should be analyzed for non-isothermal analyzing methods, obtained from the results of DTA measurements.

Crystallization peak for non-isothermal analyzing methods obtained from DTA results
A common temperature value, which cuts all the crystallization peaks obtained from analysis, is determined, and thus, the crystallization ratio is calculated with the help of the area under these peaks up to this common temperature value. In Fig. 1, T s and T f are the temperatures which crystallization begins and ends, respectively; T shows any temperature value which is used to find the crystallization ratio of the sample. A is the total area under the peak, and A T represents the area between T s and T [16].
The equation that is used to calculate the crystallization rate from the curve obtained using non-isothermal analyzing method is defined as follows,
In Johnson–Mehl–Avrami equation, β is expressed as,
where β is conversion rate, k is reaction rate constant, and n is the Avrami constant. In order to calculate the size of the crystallization, Johnson–Mehl–Avrami equation is rearranged as,
where α is the heating rate. The slope of ln[−ln(1 − β)] versus lnα gives the value of −n.
According to Xie and Gao, the Avrami parameters found from surface to inside are [16];
-
n = 1; surface nucleation and one-dimensional growth
-
n = 2; volume nucleation and one-dimensional growth
-
n = 3; volume nucleation and two-dimensional growth
-
n = 4; volume nucleation and three-dimensional growth.
Displacement in exothermic peak temperature associated with the heating rate is also used in the activation energy calculation [17]. In our study, crystallization activation energy was found by using Kissinger, Takhor and Augis–Bennett methods. All three methods have been developed by basing on the Johnson–Mehl–Avrami equation [18]. Activation energy of the samples by Kissinger method can be calculated using equation,
where E is the activation energy and R is the universal gas constant (R = 8.314 J mol−1 K) [19, 20]. According to the crystallization temperature peaks obtained from DTA curve, the slope of \( { \ln }\left( {\alpha /T_{\text{p}}^{2} } \right) \) versus 1000/T p plot is equal to −E/R.
Another method for the calculation of the activation energy is Takhor method [21]. According to this method, activation energy is given by,
As it is seen, the expressions here are same with Kissinger method. Activation energy can be calculated from the slope lnα versus 1000/T p plot. Another alternative method is Augis–Bennett method. According to this method, activation energy is given by following formula [22].
where T 0 is absolute temperature. Similarly as in the other methods, the activation energy can be calculated from the slope of ln(α/(T p – T 0)) versus 1000/T p plot. Their possible technological applications in future should be considered during the characterization of the materials. Investigating the micromechanical properties of a material is another useful method in order to realize the eligibility of the material in technology. Determining the micromechanical properties of a material is as important as determining the structural, thermal, optical, electrical and magnetic properties of the materials.
Hardness of the material is one of the most fundamental quantities in mechanical characterization. Hardness can be expressed as the resistance of the surface of a material to the applied load. Although hardness does not give basic information, it is widely used in industry to determine the production quality of materials and especially controlling the efficiency of thermal process and it is also quickly and simple applied nondestructive test. Mechanical properties of ceramic materials such as elastic modulus, yield strength, fracture toughness and hardness are important as electrical, thermal, magnetic and microstructural properties of the material [23–25]. Determination of the mechanical properties of materials plays a very important role in the improvement in the commercial applications of materials under study.
Hardness (H v), elastic modules (E), yield strength (Y) and fracture toughness (K IC) were determined to analyze the mechanical characterization of materials by using Eqs. (7–10).
Results and discussion
Crystallization kinetics and thermogravimetric analyses
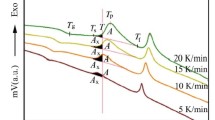
The DTA plots of the sample without Ru doping at α = 5, 10, 15, 20 K min−1 heating rates are given in Fig. 2, and at α = 5 K min−1 heating rate, two exothermic peaks appeared at 742.5 and 815 K. At this temperature region, the first peak (T p), occurred at exothermic events in BSCCO system, corresponds to Bi-2201 phase formation. The second peak between 815 and 854 K (depending on the heating rate) can be due to the formation of low-temperature impurity phases, such as Ca2PbO4 or CuO contained impurity phases [26]. The third peak, occurred at high heating rates in 850- to 900-K region, belongs to 2212 phase and consistent with the literature [27]. First and second peak values for all samples are given in Table 1.

DTA plots of Bi1.8Pb0.2Sr2CaCu2O10+δ sample for various heating rates
The slope of the line in ln[−ln(1 − β)] versus lnα graph (Fig. 3), plotted by using Ozawa equation (Eq. 3), gives the Avrami parameter. The Avrami parameters, calculated for doped and undoped samples via Ozawa equation, were found approximately as 3 (Table 1). This result is the indication of the volume nucleation and two-dimensional growth for the samples. Ru–Bi partial substitution has no effect on nucleation characteristics.

ln[−ln(1 − β)] versus ln(α) graph for the samples, obtained by Ozawa equation
First crystallization temperature was obtained in the range of 742.5 and 759.1 K for undoped Bi1.8−x Ru x Pb0.2Sr2CaCu2O10+δ sample. Taking into account the 10-K min−1 heating rate given in Table 1, the peak temperatures were determined as 752.8, 763.0 and 763.6 K for x = 0.025, 0.050 and 0.075, respectively. When the T p2 values were analyzed, a decrease in the temperature value was observed with Ru doping with respect to the undoped sample. Both cases indicate that low concentration Ru ions in the structure have an active effect at the system.
Increasing Ru and decreasing Bi concentration, causing an increase in 2201 peak temperature, were also observed as independent of nucleation. The crystallization temperature shifted to high-temperature region when the heating rate was increased from 5 to 20 K min−1. This is directly related to the nucleation rate of the sample. For slow heating rate, nucleation will increase in the sample, due to the increase in energy storage time. In contrary to this, if the heating rate is high, the energy required for nucleation will pass very fast. Thus, nucleation cross section will be reduced. This implies that energy needed for nucleation is provided at higher temperatures.
The crystallized fraction value as a function of temperature and time at 10 K min−1 heating rate is plotted in Fig. 4. From this figure, the crystallization beginning–final temperature rates were determined as 738.4–777.3, 754–777, 752.2–778 and 759.2–786 K for Ru0, Ru025, Ru050 and Ru075 samples, respectively. Bi-2201 temperature rates were found to be reduced for samples containing Ru. By taking into account the constant heating rate 10 K min−1, the time for Bi-2201 was 3.69, 3.43, 2.78 and 2.91 min for Ru0, Ru025, Ru050 and Ru075 samples, respectively. It is clearly seen that the time for nucleation was reduced with increasing doping. This situation shows us the increasing Ru concentration may reduce the amount of nucleation. In Fig. 5, the crystallization rate as a function of temperature was given for Ru–Bi partial substituted samples at 10 K min−1 heating rate. When the beginning and ending values of the peaks are analyzed, it is seen that temperature value increases with increasing Ru concentration.

Crystallized fraction as a function of temperature and time at 10 K min−1 for the samples

Crystallized rate as a function of temperature at 10 K min−1 for all samples
The activation energies for the first peak Bi-2201 of the samples were calculated by using Kissinger, Augis–Bennett and Takhor Models. Graphics and results by using these models are given in Figs. 6–8 and Table 2.

Kissinger plots for the crystallization of the samples Ru0, Ru025, Ru050 and Ru075

Takhor plots for the crystallization of the samples Ru0, Ru025, Ru050 and Ru075

Augis–Bennett plots for the crystallization of the samples Ru0, Ru025, Ru050 and Ru075
Activation energies required for Bi-2201, calculated with Kissinger, Augis–Bennett and Takhor models, are consistent with the studies in the literature [27, 28]. Activation energy for undoped sample, calculated from the slope of the line in Figs. 6–8 by using Eqs. (4)–(6) with the help of three methods, was approximately 158 kJ mol−1 higher than that of Ru025 sample. In order for crystallization, low-doped sample requires less energy by comparing to undoped sample. The activation energy values were increased by doping. Accordingly, the difference in activation energy values between the samples was reduced. The reason for the low activation energy required for nucleation can be related to high viscosity in the molten and subsequent supercooled state. Furthermore, in the Ru025 sample, having the lowest activation energy, crystallization and viscous flow were easier than other samples.
The curves given in Fig. 9 represent the thermogravimetric (T G) plots of all samples at 10 K min−1 heating rate. All data obtained from T G curves and calculation results by using these data are listed in Table 3. It is obvious from this table that the total mass gain was increased from 1.55 × 10−4 to 3.48 × 10−4 g with increasing Ru concentration at 10 K min−1 heating rate. This situation is the same for all heating rates, in general. Considering the total mass gain (Δm) and temperature difference (ΔT) ratio, a slight decrease was observed for Ru025 sample at all heating rates, but an increase with increasing Ru concentration. That is worth to say, low Ru doping and Bi deficiency caused a certain reduction in the amount of oxidation. When the relationship between the resulting mass gain and the time of acquisition was examined, it may be noted that the oxidation was faster on undoped sample, but it was slowed down by increasing partial Ru–Bi substitution oxidation. For instance, the mass gain per unit of time was 6.62 g s−1 for RuO sample at 10 K min−1, and this value was increased to 8.91 g s−1 for Ru075 sample. Since the obtained mass gain is directly related to oxidation, the oxygen gain per temperature of the samples was similarly calculated as 11.47 atom K−1 for Ru0 and 15.57 atom K−1 for Ru075. The obtained and calculated values show similar behavior at all heating rates.

TG plots of all samples under oxygen atmosphere at 10 K min−1 heating rate
XRD analyses
XRD graphs belonging to samples Ru0, Ru025, Ru050 and Ru075, prepared by using glass ceramic method and with 120 h of heat treatment at 845 °C, are given in Fig. 10. Main phase was determined as Bi1.6Pb0.4Sr2CaCu2Ox (045-0676-ICDD) from undoped Ru0 sample. According to this figure, some of the XRD peaks are belonging to Bi-2212 (045-1058-ICDD), Bi-2201 (047-0737-ICDD) phases observed in addition to the main structure, and some of them are belonging to Sr3PbO (083-1866-ICDD), SrBi4O7 (046-0752-ICDD) and Ca6Sr8Cu24O41 (048-1502-ICDD) impurity phases.

XRD patterns for all samples
It was found that all the samples have tetragonal crystal structure, and their calculated lattice parameters are given in Table 4. As it can be seen from this table, a and c parameters of the samples were decreased with Ru doping. Lattice parameter a was decreased from 5.40 to 5.38 Å, and parameter c was decreased from 30.87 to 30.78 Å, and in conjunction with these values, the related lattice volumes increased from 900.619 Å for undoped sample to 892.350 Å for Ru075 sample.
SEM analyses
To investigate the surface morphology and grain orientations of the glass samples, JEOL 7000F SEM was used. As it can be seen from SEM images in Fig. 11, grain sizes and the porosity between grains were decreased with doping. Although all samples were sintered at the same temperature, locally melting was observed for Ru075 sample. From DTA curves, a decrease in the temperature of the crystallization determined with the decreasing amount of doping. This means that the resistance of the structure to temperature decreased by doping. Namely, the crystallization was decreased by the amount of doping. Hence, flaky-like and needle grains occurred only at the undoped sample. Increase in the amount of doping prevented the formation of the needle-like structure.

SEM micrographs for all samples. a Ru0, b Ru025, c Ru050, d Ru075
Micromechanical characterization
Figure 12 shows the Vickers microhardness values as a function of the applied load for the samples. According to curves in this figure, it is understood that Vickers microhardness values of the samples increase with decreasing Ru doping and increase with the applied load. The microhardness values reach to plateau limits at about 2 N for all samples. According to the similar studies in the literature, all microhardness values must reach to plateau limit [29–32]. Beyond this limit, the load, applied to the surface of the sample, does not cause any significant changes on hardness. Reverse indentation size effect (RISE) behaviors were seen for undoped and all Ru-doped samples according to Fig. 12. Shortly, RISE behavior is defined as the increase in microhardness with applied load [33, 34]. Whereas small effect of indenter for low load and vibration effect, having a defective region in the crystal, energy loss resulting from deformation around the trace formed during indentation, cracks formed during indentation is among the reasons for the RISE behavior, this phenomenon is not fully elucidated.

Variations of microhardness with applied load for the samples
E, Y and K IC values depending on the hardness of Ru0, Ru025, Ru050 and Ru075 samples are given in Table 5. It is an expected result that, like hardness values, these values decrease with Ru addition. We can explain this as elastic modulus, in other words rigidity, is the resistivity to elastic deformation of a material. High elastic modulus means the material is rigid or elastic unit deformation caused by stress is small.
Conclusions
In this study, the effects of Ru–Bi partial substitutions on thermal, microstructural and micromechanical properties of Bi1.8−xRuxPb0.2Sr2CaCu2O10+δ glass ceramics have been investigated. Two crucial results obtained from DTA data. First, crystallization peak temperatures of undoped sample occur at lower temperatures than the other samples. On account of this situation, Ru doping causes the crystallization at higher temperature by reducing the thermal activities of ions in the structure. The activation energies of the samples for crystallization were calculated by using Kissinger, Augis–Bennett and Takhor methods. Activation energy was reduced for x = 0.025, and it was found to be increased for higher doping values. Hence, it was determined that thermodynamically more unstable samples were obtained with Ru doping. The most stable sample was the undoped Bi1.8−xRuxPb0.2Sr2CaCu2O10+δ sample. The Avrami parameters, calculated via Ozawa equation for all samples, were found approximately 3. This result is the indication of volume nucleation and two-dimensional growth. With Ru–Bi substitution, nucleation and crystallization transition temperatures of the glass materials shifted to higher temperatures and the nucleation time was reduced. These results show that the number of nucleation will decrease with Ru doping. Furthermore, with this study, it is seen that Ru doping has also an effect on oxidation increase. Mechanical characterization of the samples was obtained by Vickers microhardness test method. Microhardness increased with increasing applied load for all samples, and RISE behavior was observed. Furthermore, microhardness decreased with increasing Ru doping; accordingly, elastic modulus, yield strength and fracture toughness values decreased.
References
Omar AA, El-Shennawi AWA, El-Ghannam AR. Thermal expansion of glasses and corresponding glass–ceramics. J Mater Sci. 1991;26:6049–56.
Sitarz M, Bulat K, Wajda A, Szumera M. Direct crystallization of silicate–phosphate glasses of NaCaPO4–SiO2 system. J Therm Anal Calorim. 2013;113:1363–8.
Wers E, Oudadesse H. Thermal behaviour and excess entropy of bioactive glasses and Zn-doped glasses. J Therm Anal Calorim. 2014;115:2137–44.
Sakamoto A, Yamamoto S. Glass ceramics: engineering principles and applications. Int J Appl Glass Sci. 2010;1:237–47 (Special Issue: Special Applications).
Koralay H, Hicyilmaz O, Cavdar S, Asikuzun E, Tasci AT, Ozturk O. Effect of Zn content on microstructure and mechanical performance in Bi1.8Sr2Ca2Cu3.2−x Zn x O10+δ glass ceramic. J Mater Sci Mater Electron. 2014;25:3116–26.
Çavdar Ş, Koralay H, Altındal Ş. Effect of vanadium substitution on the dielectric properties of glass ceramic Bi-2212 superconductor. J Low Temp Phys. 2011;164:102–14.
Uzun O, Kölemen U, Çelebi S, Güçlü N. Modulus and hardness evaluation polycrystalline superconductors by dynamic microindentation technique. J Eur Ceram Soc. 2005;25:969–77.
Kölemen U, Çelebi S, Karal H, Öztürk A, Çevik U, Nezir S, Görür O. Superconducting and Vickers hardness properties of ZnO added YBCO bulk superconductors. Phys Status Solidi (b). 2004;241:274–83.
Asikuzun E, Ozturk O, Cetinkara HA, Yildirim G, Varilci A, Yılmazlar M, Terzioglu C. Vickers hardness measurements and some physical properties of Pr2O3 doped Bi-2212 superconductors. J Mater Sci Mater Electron. 2012;23:1001–10.
Matsushita K, Sakka S. Kinetic study on non-isothermal crystallization of glass by thermal analysis. J Non-Cryst Solids. 1980;741(38–39):741–6.
Prasad NS, Varma KBR. Crystallization kinetics of the LiBO2–Nb2O5 glass using differential thermal analysis. J Am Ceram Soc. 2005;88:357–61.
Araujo EB, Idalgo E, Moraes APA, Filho SAG, Filho MJ. Crystallization kinetics and thermal properties of 20Li2O–80TeO2 glass. J Mater Res Bull. 2009;44:1596–600.
Yukimitu K, Oliveira RC, Araujo EB, Moraes JCS, Avanci LH. DSC studies on crystallization mechanisms of tellurite glasses. J Thermochim Acta. 2005;426:157–61.
Davim EJC, Senos AMR, Fernandes MHV. Non-isothermal crystallization kinetics of a Si–Ca–P–Mg bioactive glass. J Therm Anal Calorim. 2014;117:643–65.
Zivanovic VD, Tosic MB, Grujic SR, Matijasevic SD, Stojanovic JN, Nikolic JD, Smiljanic SV. DTA study of the crystallization of Li2O–Nb2O5–SiO2–TiO2 glass. J Therm Anal Calorim. 2015;119:1653–61.
Xie X, Gao H. Calorimetric studies on the crystallization of Li2S–B2O3 glasses. J Non-Cryst Solids. 1998;240:166–76.
Arslan A, Koralay H, Çavdar Ş, Günen A. Crystallization kinetic studies on Bi1.75Pb0.25Sr2Ca2Cu3−x Sn x O δ glass–ceramic by using non-isothermal technique. J Non-Cryst Solids. 2012;358:1190–5.
Avrami M. Kinetics of phase changes, 1. General theory. J Chem Phys. 1939;7:1103–12.
Kissinger HE. Variation of peak temperature with heating rate in differential thermal analysis. J Res Nat Bur Stand. 1956;57:217–21.
Kissinger HE. Reaction kinetics in differential thermal analysis. Anal Chem. 1957;29:1703.
Takhor RL. Advances in nucleation and crystallization of glasses. Symposium of the Glass Division of the American Ceramic Society. Columbus; 1972. p. 166–72.
Augis JA, Bennett JD. Calculation of the Avrami parameters for heterogeneous solid state reactions using a modification of the Kissinger method. J Therm Anal. 1978;13:283–92.
Ozturk O, Erdem M, Asikuzun E, Yildiz O, Yildirim G, Varilci A, Terzioglu C. Investigation of indentation size effect (ISE) and micro-mechanical properties of Lu added Bi2Sr2CaCu2O y ceramic superconductors. J Mater Sci Mater Electron. 2013;24:230–8.
Koralay H, Cavdar S, Aksan MA. Kinetics of non-isothermal crystallization of Bi3Sr2Ca2Cu3−x Sn x O10+δ glass ceramics. Phys B Condens Matter. 2010;405:4801–5.
Cavdar S, Deniz E, Koralay H, Ozturk O, Erdem M, Gunen A. The effect of PbSe addition on the mechanical properties of Bi-2212 superconductors. J Supercond Nov Magn. 2012;25:2297–307.
Abe Y. Superconducting glass–ceramics in Bi–Sr–Ca–Cu–O: fabrication and its application. Singapore: World Scientific Publishing Co. Pte. Ltd.; 1997. p. 3–12.
Koralay H, Yakuphanoglu F, Cavdar S, Günen A, Aksu E. Crystallization kinetics of Bi1.7V0.3Sr2Ca2Cu3O x glass–ceramic. Phys B Condens Matter. 2005;355:64–71.
Aksan MA, Yakinci ME, Balci Y. Thermal analysis study of Bi2Sr2Ca2Cu3−x Er x O10+δ glass–ceramic system. J Therm Anal Calorim. 2005;81:417–23.
Safran S, Kılıç A, Asikuzun E, Kılıçarslan E, Ozturk O, Gencer A. Influence of different boron precursors on superconducting and mechanical properties of MgB2. J Mater Sci Mater Electron. 2014;25:2737–47.
Arda L, Ozturk O, Asikuzun E, Ataoglu S. Structural and mechanical properties of transition metals doped ZnMgO nanoparticles. Powder Technol. 2013;235:479–84.
Koralay H, Arslan A, Cavdar S, Ozturk O, Asikuzun E, Gunen A, Tasci AT. Structural and mechanical characterization of Bi1.75Pb0.25Sr2Ca2Cu3−x Sn x O10+y superconductor ceramics using Vickers microhardness test. J Mater Sci Mater Electron. 2013;24:4270–8.
Tosun M, Ataoglu S, Arda L, Ozturk O, Asikuzun E, Akcan D, Cakiroglu O. Structural and mechanical properties of ZnMgO nanoparticles. Mater Sci Eng A. 2014;590:416–22.
Awad R, Abou-Aly AI, Kamal M, Anas M. Mechanical properties of (Cu0.5Tl0.5)-1223 substituted by Pr. J Supercond Nov Magn. 2011;24:1947–56.
Sangwal K. On the reverse indentation size effect and microhardness measurement of solids. Mater Chem Phys. 2000;63:145–52.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ozturk, O., Gokcen, T., Cavdar, S. et al. A study on nucleation, crystallization kinetics, microstructure and mechanical properties of Ru–Bi partial substituted BSCCO glass ceramics. J Therm Anal Calorim 123, 1073–1082 (2016). https://doi.org/10.1007/s10973-015-5028-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-015-5028-8