
Abstract
[ML2]·yC2H5OH·zH2O complexes (M=CoII (1) and NiII (2), y = 4, z = 0; M=CuII (3), y = 0, z = 3; HL = N-(2-thiazolyl)-1H-benzotriazole-1-carbothioamide) were prepared, characterized (elemental analysis, TG, FT IR, UV–Vis, EPR, magnetic and conductance measurement) and tested for their antimicrobial activity against Escherichia coli and Staphylococcus aureus. Complexes 1–3 consist of a metal center having a considerable tetrahedral distortion in the xy-plane from the square planar stereochemistry MN2S2 formed by the two deprotonated benzotriazole ligands. TD-DFT calculations were carried out at B3LYP/6-31G* level of theory to understand the electronic structure and to explain the related experimental findings. Natural bond orbital analysis was performed to provide details about the electronic arrangement, type of hybridization and the nature of bonding. Coordination of HL to CoII gave rise to inactive compound, but the development of NiII and CuII complexes did not clearly change the toxicity of the free HL.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The carbothioamide derivatives and their coordination compounds [1, 2] received remarkable interest because of their biological properties as antiviral [3], antibacterial [4] and HDL-elevating properties [5]. The coordination behavior of carbothioamides toward different metal ions contributed also to a great extent to understanding of the mechanisms of interaction with enzymes and cell thiols. Generally, carbothioamides belong to thiourea group, which was recognized to possess a wide range of industrial applications as catalysts [6, 7], inhibitors [8] and for extraction of the toxic metals [9] as well as diverse biological activity [3, 10–12]. The coordinating ability of N,S-ligands permitted them to form complexes of intriguing research applications [13–15]. For example, Ni(II) complexes of thiourea derivatives were attracted much attention because of their use as models for the nickel center of enzymes [16]. Urease is a nickel-containing enzyme, which catalyzes the hydrolysis of urea microbes, higher plants and soil.
N-(2-thiazolyl)-1H-benzotriazole-1-carbothioamide (Scheme 1, HL) is a polydentate ligand having several coordination sites (benzotriazole, carbothioamide and thiazole ring). The incorporation of the biologically active heterocyclic rings such as benzotriazole and thiazole in the molecular structure of this N,S-donor derivative represents an interesting strategy to design new pharmaceutical candidates [17, 18]. Recently, we synthesized Zn(II), Pd(II) and Pt(II) complexes of HL [15] and screened them for their antitumor activity on MCF7 tumor and also against some microorganisms for their antimicrobial activity. Different coordination modes were reported. The thiazole N and C–S− donor sites of two ligand molecules are coordinated to Zn(II), while in case of Pd(II) and Pt(II), the benzotriazole N and C–S− are participated in the complex formation. Pd-L complex is triple times more toxic than cisplatin.

Synthesis of [ML2]·yC2H5OH·zH2O complexes, HL = N-(2-thiazolyl)-1H-benzotriazole-1-carbothioamide
In the present study, synthesis, spectroscopic, structural properties and biological activity evaluation of Co(II), Ni(II) and Cu(II) complexes of HL are reported (Scheme 1). The experimental studies were complemented by quantum chemical calculations at DFT/B3LYP/6-31G* level of theory. The spectral properties and electronic structure were studied by TD-DFT calculations.
Experimental
Instruments
FT IR spectra were recorded as potassium bromide pellets using a Jasco FTIR 460 plus in the range of 4000–200 cm−1. 1H NMR spectra were recorded in DMSO-d6 using Bruker 200 spectrometer at the ambient temperature. TG was performed in nitrogen atmosphere (20 mL min−1) in a platinum crucible with a heating rate of 10 °C min−1 using a Shimadzu DTG-60H simultaneous DTG/TG apparatus. A digital Jenway 4310 conductivity with a cell constant of 1.02 was used for the molar conductance study. Elemental microanalysis was performed using Elementar Vario EL III. Magnetic measurement was carried out on a Sherwood Scientific magnetic balance using Gouy method [19], and Hg[Co(SCN)4] was used as a calibrant. Electronic spectra were scanned on a Shimadzu Lambda 4B spectrophotometer in DMF. Solid X-band EPR measurements were performed at 298 K using a Bruker EMX spectrometer. The magnetic modulation frequency was 100 kHz, and the microwave power was set to 0.201 mW. The g values were obtained by referencing to a diphenylpicrylhydrazyl (DPPH) sample with g = 2.0036. The modulation amplitude was suited at 1 Gauss, while the microwave frequency was determined as 9.7 GHz.
Synthesis
Five milliliters of aqueous solution containing one mmol of Co(ClO4)2·6H2O (365 mg), Ni(ClO4)2·6H2O (366 mg) and Cu(ClO4)2·6H2O (370 mg) was mixed with 2 mmol of HL (522 mg) (supplied from Sigma chemical company) suspended in 15 mL ethanol, and then, the solution was refluxed for 3 h, where the complex was precipitated.
-
Complex 1: Color: Buff. Elemental analysis (%): calcd. C28H36CoN10O4S4: C 44.03, H 4.75, N 18.34, found C 43.91, H 4.69, N 17.84. FT IR (cm−1): 3438 ν(OH)ethanol, 2970, ν ass(CH3)ethanol, 2866, ν ss(CH3)ethanol, 2709, ν ss(CH2)ethanol, 1597, ν(C=N)thiazole + ν(C=N)Thiol, 1375, ν(N=N), 1216, (ν(C=N) + ν(C=S) + β(C–H)) II, 1035 ν(N–N), 761 δ(C=S). UV–Vis. (DMF, 10−4, nm): 265, 295, 330, 490, 620, 670. Molar Cond. (10−3 M, DMF, Ω−1 cm2 mol−1): 8.47. μ eff (μ B, 298 K): 3.44.
-
Complex 2: Color: Pale brown. Elemental analysis (%): calcd. C28H36NiN10O4S4: C 44.04, H 4.75, N 18.34, found C 43.45, H 4.22, N 17.84. FT IR (cm−1): 3444 ν(OH)ethanol, 2972, ν ass(CH3)ethanol, 2701, ν ss(CH2)ethanol, 1593, ν(C=N)thiazole + ν(C=N)Thiol, 1370, ν(N=N), 1214, (ν(C=N) + ν(C=S) + β(C–H)) II, 1038 ν(N–N), 760 δ(C=S). UV–Vis. (DMF, 10−4, nm): 245, 265, 290, 320, 390, 590. Molar Cond. (10−3 M, DMF, Ω−1 cm2 mol−1): 27.40. μ eff (μ B, 298 K): 2.22.
-
Complex 3: Color: Brick-red. Elemental analysis (%): calcd. C20H18CuN10O3S4: C 37.64, H 2.84, N 21.95, found C 37.99, H 3.04, N 21.69. FT IR (cm−1): 3434 ν(OH)water, 1598, ν(C=N)thiazole + ν(C=N)Thiol, 1231 (ν(C=N) + ν(C=S) + β(C–H)) II, 1006, ν(N–N), 792 δ(C=S). UV–Vis. (DMF, 10−4, nm): 245, 265, 320, 380. Molar Cond. (10−3 M, DMF, Ω−1 cm2 mol−1): 20.08. μ eff (μ B, 298 K): 1.04.
DFT calculations
Density functional theory (DFT) calculations were carried out by Gaussian 03 [20]. Ground state geometry optimization in the gaseous state of complexes 1–3 was carried out by B3LYP functional combined with 6-31G(d) basis set [21, 22]. The complexes were characterized as local minima through harmonic frequency analysis. Electronic transitions (in DMSO) were calculated by time-dependent DFT [23, 24]. NBO analysis and the analysis frontier molecular orbitals were performed at the same level of theory.
Antibacterial activity
The antimicrobial activities of the test samples were determined by a modified Kirby–Bauer disk diffusion method [25] under standard conditions using Mueller–Hinton agar medium (tested for composition and pH), as described by NCCLS [26]. The antimicrobial activities were carried out using culture of Staphylococcus aureus as gram-positive bacterium and Escherichia coli as gram-negative bacterium. The solution of 20 mg mL−1 of each compound (free ligand, metal complexes and standard drug tetracycline) in DMSO was prepared for testing. Centrifuged pellets of bacteria from a 24-h-old culture containing approximately 104–106 CFU mL−1 (colony forming unit) were spread on the surface of Mueller–Hinton agar plates. Then, the wells were seeded with 10 mL of prepared inocula to have 106 CFU mL−1. Petri plates were prepared by pouring 100 mL of seeded nutrient agar. DMSO (0.1 mL) alone was used as control under the same conditions for each microorganism, subtracting the diameter of inhibition zone resulting with DMSO, from that obtained in each case. The antimicrobial activities could be calculated as a mean of three replicates.
Results and discussion
The reaction of Co(ClO4)2·6H2O, Ni(ClO4)2·6H2O and Cu(ClO4)2·6H2O with two equivalents of HL in water/ethanol mixture gives buff (1), pale brown (2) and brick red (3) complexes in 1:2 (M:L) ratio. The complexes studied were elucidated using elemental analysis, TG, FT IR, EPR, UV–Vis, magnetic and conductance measurements. Lower molar conductance values were found for complexes 1–3 compared with the reported values [27] for 1:1 (65–90 Ω−1 cm2 mol−1) and 1:2 (130–170 Ω−1 cm2 mol−1) electrolytes in DMF, reflecting their non-electrolytic nature.
Vibrational assignments
The IR bands at 3439, 1561, 1503, 1352, 1006 and 906 cm−1 in the spectrum of HL are allocated for ν(NH), ν(C=N)Thiazole, (β(N–H) + β(C=C)) I, (ν(C=N) + ν(C=S) + β(C–H)) II, (ν(C–N) + ν(C–S)) III and δ(C=S) IV, respectively [15].The absence of the ν(S–H) band near 2570 cm−1 suggests the existence of HL in the –NH–C=S– form in the solid state. The triazole modes (ν(N=N) and ν(N–N)) could not be assigned owing to presence of intra-H-bond [28]. In complexes, coordination of HL to Mn+ gave rise to remarkable changes in 1530–1625 cm−1 range. A new band assigned to ν(C=N)Thiazole + ν(C=N)Thiol is observed at 1597 (1), 1593 (2) and 1598 cm−1 (3), since the anionic L while coordinated to the metal ion via C–S− generates a double bond. This has been confirmed by observing the II and IV modes of the C=S group at lower wave numbers, 1231–1215 and 792–757 cm−1. The observation of the combination of ν(C=N)Thiazole + ν(C=N)Thiol around 1595 cm−1 compared with the previously reported complexes [15] suggests the participation of thiazole ring in chelation. The ν(N=N) and ν(N–N) are observed at 1375, 1036 cm−1 for 1 and 1370, 1038 cm−1 for 2 revealing that the benzotriazole ring remains intact, while in 3 these modes are still overlapped. Hence, benzotriazole ring in 3 may be considered as the magnetic exchange pathway by occupying the axial position of CuII ion of the adjacent molecule as confirmed by the magnetic measurement (discussed later). Therefore, HL behaves as a mono-negative bidentate ligand via C–S− and N-thiazole ring.
TG
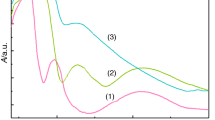
The curves of the investigated complexes reported in nitrogen atmosphere (20 mL min−1) with a heating rate of 10 °C min−1 are shown in Fig. 1. For complexes 1 and 2, the thermal decomposition of four H-bonded ethanol molecules (assigned by 1H NMR data) and two L preceded through three complicated steps at 178, 476 and 964 °C for 1 and 163, 447 and 534 °C for 2. The final residue may be considered to be metallic cobalt (observed: 6.68 %, calcd.: 7.61 %) and NiS (observed: 11.43 %, calcd.: 11.81 %) [29]. The TG curve of 3 exhibited five distinct peaks at 223, 263, 345, 633 and 919 °C. The first and second stages are accompanied by a mass loss amounts to 45.31 %. This value finds a parallelism with the calculated value (45.43 %) responsible for desorption of 3H2O of hydration and loss of two benzotriazole rings. The third, fourth and fifth thermal stages bring the total mass loss up to 77.81 % (calcd. 78.15 %) leaving carbonized CuS2 as a final residue.

TG and DTG curves of complexes (a) 1, (b) 2 and (c) 3
Electronic structure, magnetic and EPR
The electronic spectrum of HL exhibited three absorption bands at 365, 310 and 270 nm in DMF [15]. In the highest energy region, compound 1 showed two bands at 265 and 295 nm as well as a shoulder at 330 nm allocated for the internal ligand transitions. Coordination of HL via the C–S− was established by the disappearance of the band at 365 nm. The excitation energy at 330 nm proved the participation of the thiazole ring in the chelation. Complex 1 displayed three weak additional bands in the visible region at 20,408, 16,129 and 14,925 cm−1 that might be accounted for 4A2 → 4T1(P), 4A2 → 4T1(F) and 4A2 → 4T1, respectively, in a tetrahedral field [30, 31]. Generally, octahedral Co(II) complexes comprise large orbital contribution and exhibit μ eff above 4.7 μB, while the low-spin square planar complexes with only one unpaired electron and large orbital contribution may give μ eff values around 3.0 μB. The magnetic moments of tetrahedral Co(II) complexes are always greater than the spin only value via the orbital contribution [32]. The effective magnetic moment value of 3.44 μB for 1 is in the expected range of Co(II) ion in a distorted tetrahedral field [32].
The room temperature μ eff of 2.22 μB for 2 is in the expected range characteristic of square planar–tetrahedral Ni(II) complexes [33]. The percentage of tetrahedral (N t) in this mixture is found to be 45 % as calculated by N t = [100 × (μ eff)2/(3.3)2] relation (3.3 μB is the magnetic moment for ideal tetrahedral Ni(II) complexes). Like compound 1, the spectrum of 2 showed four electronic transitions at 245, 265, 290 and 320 nm. Two additional transitions were observed in 2 at 25,641 and 16,949 cm−1 assigned to LMCT (S(σ) → Ni(II)) [34] and \( d_{{\text{z}^{2} }} \to d_{{\text{x}^{2} - \text{y}^{2} }} \) (1A1g → 1B1g) [35] transitions, respectively, suggesting the predominance of the square planar stereochemistry in DMF solution.
Complex 3 showed anomalous room temperature μ eff of 1.04 μB indicating the presence of strong anti-ferromagnetic coupling between the CuII centers of the neighboring molecules [36]. The magnetic exchange interactions between the paramagnetic ions may arise from the direct interaction and/or super-exchange depending upon the distance of Cu···Cu separation. In the super-exchange process, the π orbital of a bridged group overlaps with both copper atoms, where the unpaired spins may migrate to the bridged groups and become paired. The direct interaction of the adjacent copper(II) ions is mainly due to the formation of a δ-bond, \( d_{{\text{x}^{2} - \text{y}^{2} }} \) orbital overlap. In complex 3, the magnetic exchange pathway is considered to proceed through the uncoordinated benzotriazole moiety, which can occupy the axial position of the CuII center of the neighboring molecule in the solid state to form a Jahn–Teller elongated octahedral stereochemistry. In other words, the mononuclear units are not magnetically isolated. The solution electronic spectrum (in DMF) of 3 showed five electronic transitions at 245, 265, 320, 380 (26,315 cm−1) and 440 nm (22,727 cm−1). The internal ligand transitions are established as in complexes 1 and 2. The band at 380 nm is ascribed to LMCT S(σ) → Cu(II) [37], while the shoulder at 440 nm may be attributed to the pyridyl N(π) → Cu(II) transition [38].
Room temperature EPR spectrum on the powdered sample of complex 3 was recorded at X-band frequency. As shown in Fig. 2, the spectrum of 3 is axial with two well-defined g|| = 2.14 (A|| = 132 × 10−4 cm−1) and g ⊥ = 2.06 values and g ∥ > g ⊥ > g e (2.0023), suggesting that the \( d_{{\text{x}^{2} - \text{y}^{2} }} \) orbital is the most populated ground state. The stereochemistry around the metal center could be estimated from the f value (g ∥/A ∥), where the values in the 105–135 cm range are recognized for the regular square planar geometry and that higher than 135 cm indicates the presence of a tetrahedrally distorted structure [39]. For 3, the \( {\text{g}}_{\parallel } /{\text{A}}_{\parallel } \) (162 cm) value indicates the presence of a considerable tetrahedral distortion in the xy-plane from the square planar geometry. The anisotropic spectrum of 3 is similar to those previously reported for distorted tetrahedral-based coordination polyhedral [40, 41]. No band corresponding to M s = 2 transition was observed in the solid spectrum of 3 ruling out the possibility of Cu–Cu interaction. The G factor, defined as G = (g ∥ − g e)/(g ⊥ − g e) for the axial spectrum [42, 43], is <4, indicating the presence of strong exchange interaction [21] between the copper molecules in the solid state.

EPR spectrum of complex 3
DFT/TD-DFT
To obtain an insight into the spectral properties and electronic structure of the complexes studied here, time-dependent density functional theory calculations were carried on the optimized structures at DFT/B3LYP/6-31G* level of theory. Before calculating the electronic structures of the complexes, their geometries should be characterized as local minima through the harmonic frequency analysis. The fully optimized geometries of [ML2]·4C2H5OH (M=Co (1) and Ni (2)) and [CuL2]·3H2O (3) as well as the numbering of atoms are shown in Fig. 3. Selected bond distances and angles are listed in Table 1. Complexes 1–3 consist of metal center having a considerable tetrahedral distortion in the xy-plane from the square planar geometry MN2S2 formed by two deprotonated ligand molecules. The angles around the metal center increase from Ni to Co and then Cu, and consequently, an elongation in the M–N and M–S bond lengths is reported. Table 1 summarizes the details of H-bond interactions. For complexes 1 and 2, four ethanol molecules interact with the mononuclear unit through five H-bonds of different strengths, 2.013–2.417 Å and 121.9°–164.8° for 1 and 2.018–2.394 Å and 122.0°–162.9° for 2. It is well known that linear H-bonds and those that approach linearity are usually stronger than the bonds where the X–H···Y angle (X and Y are electronegative elements) is far from 180° [44]. Thus, the strongest H-bond in 2 is N12···H48–O49 (2.189 Å), since its angle of interaction is 162.9°. Similarly, four intermolecular H-bonds are observed in the optimized structure of 3 coming from the interactions of three water molecules with the coordination compound. The N12···H47–O48 (163.0°) is stronger than the other interactions, because this interaction approaches more linearity.

Local minimum structures of complexes (a) 1, (b) 2, and (c) 3 obtained at the DFT/B3LYP/6-31G(d) level of theory
The nature of the electronic transitions observed in the spectra of the complexes studied has been assigned through the calculation of the lowest 30 singlet spin-allowed excitation states (Fig. 4). The calculated spectrum of 1 showed three major transitions at 462, 427 and 350 nm assigned to H(β) → L(β), H-1(β) → L+1(β) and H-3(α) → L(α)/H-3(β) → L(β) (H: HOMO; L: LUMO). The transition energy at 462 nm has a ground state composed of Co \( d_{{z^{2} }} \) character, whereas the excitation state is of d orbital forming a d–d transition (Table S1†). Four transitions are observed in the stimulated spectrum of complex 2 at 904, 460, 361 and 319 nm related to H → L/L + 2, H → L+1, H-3 → L and H-5 → L transitions (Fig. 5). HOMO is composed of Ni \( d_{{\text{z}^{2} }} \) nature with contribution from the π system of the benzotriazole rings. The two LUMOs (LUMO and LUMO + 1) are formed from Ni \( d_{{\text{x}^{2} - \text{y}^{2} }} \) and benzotriazole π* orbitals and are lying close to HOMO with a small energy gap (0.04 eV). Therefore, the bands at 904 and 460 nm are close to \( d_{{\text{z}^{2} }} \to d_{{\text{x}^{2} - \text{y}^{2} }} \), with a contribution from \( \pi \left( {\text{benzotriazole}} \right) \to d_{{\text{x}^{2} - \text{y}^{2} }} \) LMCT. HOMO-3 shows predominant character of Ni d xz or d yz with a little contribution from the C–S group. Thus, the excitation energy at 361 nm may be assigned to \( d_{\text{xz}} /d_{\text{yz}} \to d_{{\text{x}^{2} - \text{y}^{2} }} \) and S(σ) → Ni LMCT transitions. The highest energy band at 319 nm has a π–π* character of the thiazole moiety.

Theoretical TD-DFT electronic absorption spectra of complexes 1–3 calculated by DFT/B3LYP/6-31G(d)

Theoretical electronic absorption transitions of complex 2
The spectrum of 3 displayed five transitions at 514, 426, 385, 376 and 347 nm with oscillator strengths of 0.0186, 0.0326, 0.0672, 0.1605 and 0.3032. These bands are arising from β-spin H/H-1 → L, H-2/H-4 → L, H → L+2, H-4 → L/H → L+2 and H-1 → L+2 transitions. HOMO and HOMO-1 orbitals show primarily 3d character with a contribution from the heterocyclic rings. HOMO-2, which is lower than HOMO by 0.48 eV, results from the π system of benzotriazole ring and C–S group (Table S2†). The LUMO orbital with β-spin is partially of Cu \( d_{{\text{x}^{2} - \text{y}^{2} }} \). Hence, the lowest energy band at 514 nm is created from \( d_{\text{xz}} /d_{\text{yz}} \to d_{{\text{x}^{2} - \text{y}^{2} }} \) and LMCT transitions. The band at 426 nm is assigned to LMCT character, originating from the heterocyclic rings as assigned in the experimental findings.
Natural bond orbital (NBO) analysis
The electronic arrangement of the metal center and natural atomic charge of the metal ion in the reported complexes are presented in Table 2. According to NBO [23, 45], the natural charge on the metal atom is reduced significantly from the formal charge +2 as a consequence of electron density donation from the coordination sphere active centers. In complexes 1 and 2, the metal atom forms two sigma bonds with two sulfur atoms, while the two M–N bonds can be described as donation of electron density from a lone-pair (LP) orbital on each nitrogen atom to M MOs. The σ(Co–S15) bond is formed from sp 2.98 d 1.83 hybrid on the cobalt atom (which is the mixture of 17.11 % s, 51.05 % p and 31.79 % d atomic orbitals) and sp 4.76 d 0.01 hybrid on the sulfur atom (17.33 % s, 82.50 % p and 0.17 % d) and is polarized with about 81.20 % of electron density concentrated on the S15 atom. On the other hand, the σ(Co–S30) bond is created from sp 3.14 d 2.36 hybrid on Co and sp 5.89 d 0.01 hybrid on S30 atom. For 2, the Ni–S15 bond (σ(Ni–S15) = 0.899 sp 3.16 d 2.03 (Ni) + 0.436 sp 5.31 d 0.01 (S15)) is longer than Ni–S30 bond (σ(Ni–S30) = 0.458 sp 3.31 d 2.44 (Ni) + 0.888 sp 5.74 d 0.02 (S30)) owing to the increase in the population of the d orbital of the Ni atom and p orbital of the S30 atom. For 3, the interaction between the donor sites and metal ions can be considered as electron donation from the N and S atoms to the copper MOs. Table 3 lists the selected values of the E 2 values between donor–acceptor orbitals in the studied complexes (ML bonds and H-bond interactions). The larger the E 2 value, the more intensive is the interaction between electron donors and electron acceptors.
Antibacterial activity
The antibacterial activities of HL and its complexes were studied using two microbes, S. aureus and E. coli, and compared to tetracycline used as a standard. Preliminary screening was carried out at 20 mg mL−1. It was found that HL reduced the metabolic growth of the investigated bacteria to different extent and showed better toxicity against S. aureus compared with E. coli. The variation in the antimicrobial activity of HL against the two microorganisms depends either on the impermeability of the cells of the microbes or on differences in ribosome of microbial cells. The diameter of the inhibition zone of the complexes studied indicated that the antimicrobial activity of HL is affected by type of the divalent cation. Coordination of HL to Co(II) gave rise to inactive compound, but the formation of compounds 2 and 3 did markedly alter the toxicity. The inactivity of 1 may be interpreted in terms of low lipophilicity, where the penetration of the complex through the lipid membrane is decreased, and hence, they cannot block or inhibit the growth of the microorganism. In absolute terms, IC50 value of 0.084 (2) and 0.125 (3) µmol mL−1 (3) (equivalent to 64 (2) and 80 (3) µg mL−1) was determined against S. aureus. Thus, complex 2 has higher toxicity than compound 3. In comparison with the previously published complexes (ZnII, PdII and PtII) with HL [15], it is necessary to confirm that the highest activity of [PdL(EtOH)2]·Cl [15] may be recognized to its ionic nature compared with the non-electrolytic behavior of [ZnL2]·4EtOH, [PtL(EtOH)Cl] and complexes 2 and 3 studied here.
Conclusions
Co(II), Ni(II) and Cu(II) complexes of N-(2-thiazolyl)-1H-benzotriazole-1-carbothioamide were synthesized in good yields, characterized and tested for their antibacterial activity against E. coli and S. aureus. The experimental studies were complemented by quantum chemical calculations at DFT/B3LYP/6-31G* level of theory. Coordination of HL to Co(II) gave rise to inactive compound, but the formation of compounds 2 and 3 did markedly alter the toxicity. The optimized structures consist of metal center having a considerable tetrahedral distortion in the xy-plane from the square planar geometry MN2S2 formed by the two deprotonated ligand molecules. The CuII complex showed anomalous room temperature owing to presence of strong anti-ferromagnetic coupling between the CuII centers of the neighboring molecules through the uncoordinated benzotriazole moiety, which can occupy the axial position of the metal center of the neighboring molecule in the solid state. Also, the room temperature μ eff of 2.22 μB for Ni(II) complex is in the expected range characteristic of square planar–tetrahedral Ni(II) complexes. NBO analysis reveals that the strong coordination bonds results from donation of electron density from a lone-pair orbital on sulfur atoms to the acceptor metal MOs.
References
Garoufis A, Hadjikakou SK, Hadjiliadis N. Palladium coordination compounds as anti-viral, anti-fungal, anti-microbial and anti-tumor agents. Coord Chem Rev. 2009;253:1384–97.
Campana M, Laborie C, Barbier G, Assan R, Milcent R. Synthesis and cytotoxic activity on islets of Langerhans of benzamide thiosemicarbazone derivatives. Eur J Med Chem. 1991;26:273–8.
Hunter R, Younis Y, Muhanji CI, Curtin T, Naidoo KJ, Petersen M, Bailey CM, Basavapathruni A, Anderson KS. C-2-Aryl O-substituted HI-236 derivatives as non-nucleoside HIV-1 reverse-transcriptase inhibitors. Bioorg Med Chem. 2008;16:10270–80.
Seth PP, Ranken R, Robinson DE, Osgood SA, Risen LM, Rodgers EL, Migawa MT, Jefferson EA, Swayze EE. Aryl urea analogs with broad-spectrum antibacterial activity. Bioorg Med Chem Lett. 2004;14:5569–72.
Lee J, Lee J, Kang M, Shin MY, Kim JM, Kang SU, Lim JO, Choi HK, Suh YG, Park HG, Oh U, Kim HD, Park YH, Ha HJ, Kim YH, Toth A, Tran R, Pearce LV, Lundberg DJ, Blumberg PM. N-(3-Acyloxy-2-benzylpropyl)-N′-[4-(methylsulfonylamino)benzyl]thiourea analogues: novel potent and high affinity antagonists and partial antagonists of the vanilloid receptor. J Med Chem. 2003;46:3116–26.
Fuerst DE, Jacobsen EN. Thiourea-catalyzed enantioselective cyanosilylation of ketones. J Am Chem Soc. 2005;127:8964.
Wenzel AG, Jacobsen EN. Asymmetric catalytic Mannich reactions catalyzed by urea derivatives: enantioselective synthesis of β-aryl-β-amino acids. J Am Chem Soc. 2002;124:12964–5.
Khaled KF. Experimental and atomistic simulation studies of corrosion inhibition of copper by a new benzotriazole derivative in acid medium. Electrochim Acta. 2009;54:4345–52.
Fontás C, Hidalgo M, Salvadó V, Anticó E. Selective recovery and preconcentration of mercury with benzoylthiourea-solid supported liquid membrane system. Anal Chim Acta. 2005;547:255–61.
Rodrìguez-Fernandez E, Manzano JL, Benito JJ, Hermosa R, Monte E, Criado JJ. Thiourea, triazole and thiadiazine compounds and their metal complexes as antifungal agents. J Inorg Biochem. 2005;99:1558–72.
Venkatachalam TK, Sudbeck E, Uckun FM. Structural influence on the solid state intermolecular hydrogen bonding of substituted thioureas. J Mol Struct. 2005;751:41–54.
Li HQ, Yan T, Yang Y, Shi L, Zhou CF, Zhu HL. Synthesis and structure–activity relationships of N-benzyl-N-(X-2-hydroxybenzyl)-N′-phenylureas and thioureas as antitumor agents. Bioorg Med Chem. 2010;18:305–13.
Bombicz P, Mutikainen I, Krunks M, Leskelä T, Madarász J, Niinistö L. Synthesis, vibrational spectra and X-ray structures of copper(I) thiourea complexes. Inorg Chim Acta. 2004;357:513–25.
Biswas S, Yap GPA, Dey K. Reaction of diacetylmonoxime with morpholine N-thiohydrazide in the absence and in presence of a metal ion: facile synthesis of a thiadiazole derivative with non-bonded S···S interaction. Polyhedron. 2009;28:3094–100.
Mansour AM, Mohamed MF. Complexes of N-(2-thiazolyl)-1H-benzotriazole-1-carbothioamide with Pd(II), Pt(II), and Zn(II): Spectral, DFT, cytotoxicity and anti-angiogenic effect on MCF-7 cell line. Inorg Chim Acta. 2014;423:373–83.
Halcrow MA, Christou G. Biomimetic chemistry of nickel. Chem Rev. 1994;94:2421–81.
Dubey A, Srivastava SK, Srivastava SD. Conventional and microwave assisted synthesis of 2-oxo-4-substituted aryl-azetidine derivatives of benzotriazole: a new class of biological compounds. Bioorg Med Chem Lett. 2011;21:569–73.
Change CK, Myoung SK, Ward B. Cytochrome c oxidase models. A magnetically coupled heme–copper complex. J Chem Soc Chem Commun. 1982;13:716–9.
Mansour AM. Selective coordination ability of sulfamethazine Schiff-base ligand towards copper(II): molecular structures, spectral and SAR study. Spectrochim Acta A. 2014;123:257–66.
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2003) Gaussian 03, Revision B.04. Gaussian, Inc., Pittsburgh.
Mansour AM, Shehab OR. Trapping of muscle relaxant methocarbamol degradation product by complexation with copper(II) ion: spectroscopic and quantum chemical studies. Spectrochim Acta A. 2014;128:263–71.
Abdel-Ghani NT, Mansour AM. 2-[(1H-Benzimidazol-2-ylmethyl)-amino]-benzoic acid methyl ester: crystal structure, DFT calculations and biological activity evaluation. Spectrochim Acta A. 2011;81:754–63.
Mansour AM. Synthesis, spectroscopic, electrochemical, DFT and SAR studies of nifuroxazide complexes with Pd(II), Pt(II) and Ru(II). Polyhedron. 2014;78:10–7.
Mansour AM, Mohamed RR. Sulfamethazine copper(II) complexes as antimicrobial thermal stabilizers and co-stabilizers for rigid PVC: spectroscopic, thermal, and DFT studies. RSC Adv. 2015;5:5415–23.
Abdel Ghani NT, Mansour AM. Novel palladium(II) and platinum(II) complexes with 1H-benzimidazol-2-ylmethyl-N-(4-bromo-phenyl)-amine: structural studies and anticancer activity. Eur J Med Chem. 2012;47:399–411.
National Committee for Clinical Laboratory Standards, NCCLS Approval Standard Document M2-A7, Villanova, PA, 2000.
Mansour AM. Crystal structure, DFT, spectroscopic and biological activity evaluation of analgin complexes with Co(II), Ni(II) and Cu(II). Dalton Trans. 2014;43:15950–8.
Abdel-Ghani NT, Mansour AM. Novel Pd(II) and Pt(II) complexes of N, N-donor benzimidazole ligand: synthesis, spectral, electrochemical, DFT studies and evaluation of biological activity. Inorg Chim Acta. 2011;373:249–58.
El Metwally NM, Arafa R, El-Ayaan U. Molecular modeling, spectral, and biological studies of 4-formylpyridine-4N-(2-pyridyl) thiosemicarbazone (HFPTS) and its Mn(II), Fe(III), Co(II), Ni(II), Cu(II), Cd(II), Hg(II), and UO2(II) complexes. J Therm Anal Calorim. 2014;115(3):2357.
El-Asmy AA, El-Sonbati AZ, Ba-Issa AA. Synthesis and properties of 7-formyl-8-hydroxyquinoline and its transition metal complexes. Trans Met Chem. 1990;15:222–5.
Srivastava AK, Rana VB, Mohan M. Stereochemistry of some transition metal complexes of 1-phenyl-0-hydroxy-4-benzamidothiosemicarbazone. J Inorg Nucl Chem. 1974;36:2118–22.
Yamada S. Recent aspects of the stereochemistry of Schiff-base-metal complexes. Coord Chem Rev. 1966;1:415–37.
Al-Awadi N, Shuaib NM, El-Dissouky A. Synthesis and spectroscopic characterization of nickel(II) complexes of 1-benzotriazol-1-yl-[(p-X-phenyl)hydrazone]propan-2-one. Spectrochim Acta Part A. 2006;65:36–43.
Sankaraperumal A, Karthikeyan J, Nityananda-Shetty A, Lakshmisundaram R. Nickel(II) complex of p-[N, N-bis(2-chloroethyl)amino]benzaldehyde-4-methyl thiosemicarbazone: synthesis, structural characterization and biological application. Polyhedron. 2013;50:264–9.
Singh M, Pandey AK, Butcher RJ, Singh NK. Syntheses and X-ray crystallographic studies of [Ni(en)2(pot)2]0.5CHCl3 and [Ni(en)2](3-pytol)2. Polyhedron. 2009;28:461–6.
Kato M, Jonassen HB, Fanning JC. Copper(II) complexes with subnormal magnetic moments. Chem Rev. 1964;64:99–128.
Dhara PK, Pramanik S, Lu T, Drew MGB, Chattopadhyay P. Copper(II) complexes of new tetradentate NSNO pyridylthioazophenol ligands: synthesis, spectral characterization and crystal structure. Polyhedron. 2004;23:2457–64.
Sreekanth A, Kurup MRP. Structural and spectral studies on four coordinate copper(II) complexes of 2-benzoylpyridine N(4), N(4)-(butane-1,4-diyl)thiosemicarbazone. Polyhedron. 2003;22:3321–32.
Sundaravel K, Suresh E, Palaniandavar M. Synthesis, structures, spectral and electrochemical properties of copper(II) complexes of sterically hindered Schiff base ligands. Inorg Chim Acta. 2009;362:199–207.
Rybak-Akimova EV, Nazarenko AY, Krieger PW, Herrera AM, Tarasov VV, Chen L, Robinson PD. Synthesis, characterization, redox properties, and representative X-ray structures of four- and five-coordinate copper(II) complexes with polydentate aminopyridine ligands. Inorg Chim Acta. 2001;324:1–15.
Reena TA, Prathapachandra-Kurup MR. Copper(II) complexes derived from di-2-pyridyl ketone-N4-phenyl-3-semicarbazone: synthesis and spectral studies. Spectrochim Acta A. 2010;76:322–7.
Rudley RJ, Hathaway BJ. Single-crystal electronic and e.s.r. spectra of bis(aquo)monoacetylacetonatocopper(II) picrate. J Chem Soc A. 1970; 1725–1728.
Hathaway BJ, Billing DE. The electronic properties and stereochemistry of mono-nuclear complexes of the copper(II) ion. Coord Chem Rev. 1970;5:143–207.
Abdel-Ghani NT, Mansour AM. Molecular structures of antitumor active Pd(II) and Pt(II) complexes of N, N-donor benzimidazole methyl ester. J Coord Chem. 2012;65(5):763–79.
Reed AE, Curtiuss LA, Weinhold F. Intermolecular interactions from a natural bond orbital, donor–acceptor viewpoint. Chem Rev. 1988;88:899–926.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mansour, A.M. Thermal, spectral, DFT and biological activity evaluation of Co(II), Ni(II) and Cu(II) complexes of N,S-chelated benzotriazole ligand. J Therm Anal Calorim 123, 571–581 (2016). https://doi.org/10.1007/s10973-015-4978-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-015-4978-1