
Abstract
Non-isothermal crystallization kinetics of the GeTe4 chalcogenide glass was studied in dependence on particle size. Complexity of the obtained DSC data was treated by means of the Fraser–Suzuki deconvolution, and the particular crystallization sub-processes were identified and described in terms of the JMA(2) and AC kinetic models. Bulk as-prepared GeTe4 samples, on the other hand, exhibited simple zero-order (F0) crystallization kinetics. The marked difference between the powder and bulk crystallization mechanisms was explained based on the mechanically induced defects and heterogeneities, which surrogate/accelerate the primary nucleation process. This concept also accounts for the remarkable stability of the studied telluride glass. Precipitation of Te followed by second-stage GeTe crystal growth was confirmed by XRD for all of the applied experimental conditions. The dominant influence of the nucleation process on the consequent crystallization kinetics is thereby implicated. Infrared microscopy was used to confirm the existence of the particular crystallization mechanism.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
During recent decades, research into chalcogenide glasses has been primarily motivated by their optical properties in the infrared region. Examples include infrared detectors and moldable infrared optics such as lenses, infrared optical fibers or planar waveguides [1–3]. Both selenide and telluride glassy optical fibers have shown their potential for mid-IR sensing in many fields of application. Applications of selenide glasses are, however, limited to a 2–16 μm transparency window. On the other hand, telluride glass fibers due to their heavier atoms can transmit light further into the infrared region (up to 28 μm) [4]. This expansion of the transmittance region was successfully utilized in various hi-tech applications, ranging from environmental and medicinal devices to usage within the framework of the Darwin mission searching for extraterrestrial life (capture of the 15 μm CO2 band was of key importance) [5].
Great potential associated with the extended transmittance window of telluride glasses has led to intensive research in this area during the last decade. Various Te-rich chalcogenide systems were explored, and numerous materials have been tested; the Ge–Se–Te system (Te-rich compositions along the GeTe4–GeSe4 pseudo-binary line) belongs to the most promising ones in this regard [6]. Although an intensive exploration of this system has taken place for several years, very little attention has been paid to the parental GeTe4 glass.
The limited number of actually utilizable pure telluride systems is related to the main disadvantage of Te glasses—their higher tendency to crystallize is caused by the metallic behavior of Te. For telluride fibers, a major difficulty in the fabrication is to avoid any crystallization phenomena of the glass in the bulk as well as on its surface. All stages and process steps need to be controlled and optimized in this respect so that high-quality stable glasses are obtained. Furthermore, impurity levels of the starting products as well as of the final fibers require careful control; during the drawing step, the contamination by water must also be avoided by using a dry drawing atmosphere, whereas the contact with oxygen in air should also be avoided by Ar protection [6]. This is the reason why knowledge of crystallization kinetics determined under various conditions is so crucial for the successful utilization of Te-based infrared glasses.
In the present article, the non-isothermal crystallization kinetics of the GeTe4 infrared glass will be investigated in dependence on various experimental conditions (heating rate, sample form, particle size). The crystallization mechanisms occurring will be discussed in connection with the structural ordering of the formed crystalline phases.
Theory
Crystallization kinetics is usually studied by differential scanning calorimetry (DSC) according to the following equation [7]:
where Φ is the measured heat flow, ΔH is the crystallization enthalpy, A is the pre-exponential factor, E is the apparent activation energy of the process, R is the universal gas constant, T is the temperature and f(α) stands for an expression of a kinetic model with α being conversion.
In the first step of kinetic analysis, the apparent activation energy of crystallization E is determined. In the present work, we will use the Kissinger [8], Friedman [9] and modified Kissinger–Akahira–Sunose (KAS) [10] methods. The original Kissinger method is only applicable under non-isothermal conditions and is based on the shift of the maximum of the crystallization peak T p with heating rate q + according to the following equation:
The original Kissinger method utilizes the maximum rate of conversion, that is, a physically driven action independent of most peak distortion effects [11], which is the main advantage of this method. Correspondingly, the disadvantage of this method is that it provides information only for the dominant (sharpest) crystallization peak.
Apparent activation energy in dependence on degree of conversion can be obtained by using one of the isoconversional methods. Differential isoconversional Friedman’s method and integral isoconversional modified KAS method are utilized to calculate the apparent activation energy for various degrees of conversion according to the following equations:
where \(\Phi _{\alpha }\) and T α are the specific heat flow and temperature corresponding to certain chosen values of conversion α. The experimental data are again obtained from crystallization curves measured at different heating rates and are plotted for each value of α separately. In this way, eventually, the dependence of activation energy E on the degree of conversion α is obtained.
In the second step of kinetic analysis, an appropriate kinetic model for the description of crystallization peaks is determined. An algorithm based on the shape of so-called characteristic kinetic functions z(α) and y(α) was developed for this purpose [12, 13]. For non-isothermal conditions, these functions are obtained by the following transformation of experimental data:
Determination of the most suitable kinetic model utilizes the values of α corresponding to the maxima of the characteristic functions as well as the overall shape of the functions [12]. With regard to crystallization from the glassy state, one of the two following models is usually applied: the physically meaningful nucleation–growth Johnson-Mehl-Avrami (JMA) model [14–17] and the semiempirical autocatalytic Šesták–Berggren (AC) model [7].
The JMA model is a one-parameter model and can be expressed through the following equation:
where m is the parameter reflecting nucleation and crystal growth mechanisms, as well as the crystal morphology. The value of kinetic parameter m can then be calculated either from the conversion corresponding to the maximum of the y(α) function α max,y (according to Málek [13]) or based on the double logarithm function suggested by Šesták [7].
The autocatalytic AC model is usually the second in choice, as the description by this model is semiempirical and therefore of lower worthiness. It is a usual and obvious practice to check the applicability of the physically meaningful JMA model first; the empirical AC model is only applied in cases when the JMA equation cannot be used. The phenomenological basis of the AC model is, however, also its advantage over the JMA model, as it provides higher flexibility in kinetic description. The AC model can be expressed through the following equation:
Experimental
The GeTe4 glass was prepared by the melt quenching technique. Appropriate amounts of pure elements (5N, Sigma Aldrich) were inserted into a fused silica ampoule; the ampoule was degassed and sealed afterward. The batched ampoule was annealed at 950 °C for 24 h, and then, the ampoule was quenched in cold water. The composition of the glass was verified using a JEOL JSM-5500LV electron microscope with an energy-dispersive X-ray (EDX) microanalyzer (IXRF Systems, detector GRESHAM Sirius 10). The prepared glass was ground and sieved so that the following powder fractions were obtained: 0–20, 20–50, 50–125, 125–180, 180–250, 250–300 and 300–500 μm. Small pieces of glass broken off the main glass ingot were used as “bulk” samples (the main goal was to choose samples with the minimum number of mechanical defects and heterogeneities, which could serve as nucleation/crystallization centers). The bulk samples were assigned an average size d aver = 1 mm for further comparisons. The amorphous character of the GeTe4 glass was confirmed by XRD (see Fig. 1); homogeneity of the glass was verified from the position of T g (glass transition temperature), measured for several pieces of bulk glass taken randomly from the freshly opened ampule.

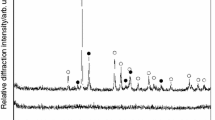
Diffraction patterns of amorphous (lower pattern) and crystallized (upper pattern) GeTe4. The particular diffraction lines corresponding to Te and GeTe are indicated
The crystallization behavior was studied using a Q2000 heat-flow DSC (TA Instruments) equipped with an autosampler, an RCS90 cooling accessory and T-zero technology. The instrument was calibrated using In, Zn and H2O; dry N2 was used as purge gas at a flow rate of 50 cm3 min−1. Fresh zero-line calibration was performed before the measurements. A thin layer of the powder was spread on the bottom of the aluminum pans to improve thermal contact. The masses of the samples varied between 8 and 10 mg. During the DSC measurements, the sample was first equilibrated at 100 °C and then heated to 300 °C (above melting point) at various defined heating rates. The applied heating rates were: 0.5, 1, 2, 3, 5, 7, 10, 15, 20 and 30 °C min−1. The XRD diffractogram of the crystallized sample is shown in Fig. 1; classification of the particular diffraction peaks is indicated. As is apparent, the hexagonal tellurium [P3121, a = 4.4572(4), b = 4.4572(4), c = 5.9290(5)] is the main crystalline phase, supplemented by rhombohedral GeTe [R3m, a = 8.3280(6), b = 8.3280(6), c = 10.690(8)]. XRD analysis of amorphous and crystalline samples was performed using a Bruker AXS diffractometer D8 Advance equipped with a horizontal goniometer and scintillation counter utilizing CuKα radiation. In order to examine development of the crystallization process, an infrared microscope Olympus BX51 equipped with XM10 camera was used in the reflection mode.
Acquisition of the DSC crystallization peak data was achieved using the cubic spline interpolation of the DSC thermokinetic background. Very good reproducibility of the experimental data was achieved. In the second part of the article, the deconvolution of the previously identified complex crystallization process will be performed. The modified Fraser–Suzuki deconvolution [18] will be used in this regard. The main advantage of this recently developed methodology is that it provides a physically meaningful separation of the overlapping kinetic processes. The deconvolution is done according to the following equation:
where a 0, a 1, a 2 and a 3 are the parameters corresponding to the amplitude, position, half-width and asymmetry of the curve, respectively.
Results and discussion
Primary kinetic analysis
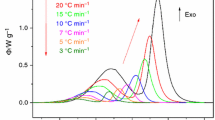
Differential scanning calorimetry was used to obtain eight datasets (corresponding to eight particle size fractions), where each dataset contains 10 crystallization heating scans performed at various heating rates. In Fig. 2, graphs for three chosen particle size fractions are shown—only data corresponding to six selected heating rates are displayed in each graph for better clarity (complete set of all 80 DSC curves can be accessed via the Supplemental material 1). In Fig. 2a, data corresponding to the 0–20 μm powder fraction are shown; the peaks are uniform, well resolved, showing negative asymmetry and single-peak-like behavior. Similar sharp and uniform peaks also occurred for the second fine powder fraction (20–50 μm), although a small peak tail manifested at the high-α side. Starting with the 125–180 μm powder, a complex crystallization behavior occurs—see Fig. 2b. The presence of two overlapping processes partially disappears in the case of the highest heating rates and converts into a shoulder at the low-α side (indicating domination of the second process). The ratio between the magnitudes of the two overlapped crystallization peaks shifts in favor of the second one also with the increasing particle size (see the Supplemental material 1). Lastly, completely different behavior can be observed in case of the bulk samples (Fig. 2c)—note the similar onset edge and the sharp decrease of the high-α peak side.

DSC curves measured for three chosen GeTe4 particle size fractions: 0–20 μm (a), 125–180 μm (b) and bulk (c). For better clarity, only data corresponding to certain selected heating rates are displayed in each graph. d The overall crystallization enthalpy ΔH evaluated in dependence on heating rate and averaged particle size
Despite the large variability of the crystallization kinetics occurring for the GeTe4 glass, the crystallization enthalpy is within the experimental error and similar for all of the measurements performed for different particle size fractions at different heating rates. In Fig. 2d, the crystallization enthalpy ΔH evaluated from all 80 measurements is plotted in dependence on heating rate q + and average particle size d aver; the averaged value of crystallization enthalpy is ΔH = 38.9 ± 2.8 J g−1 (22.7 ± 1.6 kJ mol−1). Invariability of ΔH with respect to q + and d aver indicates that the marked changes of crystallization kinetics are associated with the manifestation of different crystal growth mechanisms rather than with a change of the Te/GeTe ratio within the formed crystallites. This was further confirmed by XRD measurements performed for the crystallized 0–20 μm, 125–180 μm and bulk fractions, where no changes in the diffractograms appeared with d aver.
In Fig. 3, the primary kinetic evaluations are shown. All of the applied methodologies were derived for a single kinetic peak; nonetheless, as will be demonstrated in the following paragraphs, the application of these methods in the case of complex processes is not meaningless and can provide valuable kinetic information. In Fig. 3a, the Kissinger plot with all of the studied particle size fractions is shown. Good linearity of the displayed dependences indicates that the major crystallization mechanisms characteristic for each particular powder fraction does not change with heating rate, i.e., for each particular powder fraction, there is only one major crystallization mechanism associated with T p. Furthermore, the distribution of the particular dependences with respect to the temperature axis indicates that surface crystallization dominates in the case of GeTe4 glass [19, 20]. In Fig. 3b, the apparent activation energies evaluated by the Kissinger, KAS and Friedman methods are shown in dependence on the averaged particle size d aver. As can be seen, all three methods provide very similar results. The indifference of E with respect to d aver (E changes by ~10–15 % within the whole particle size range) confirms the uniform identity of the formed crystalline phases as well as the fact that all of the manifesting crystallization mechanisms have very similar apparent activation energies.

a Kissinger plot constructed for all of the studied GeTe4 particle size fractions. b Comparison of apparent activation energies determined for the GeTe4 glass according to Kissinger, Friedman and KAS methods in dependence on average particle size. Bulk samples were assigned d aver = 1 mm. c Particle size dependence of the characteristic kinetic functions maxima α max,z and α max,y for the GeTe4 glass. Bulk samples were assigned d aver = 1 mm. Dashed line corresponds to the theoretical “fingerprint” α max,z = 0.632 value characteristic for the JMA model. d Characteristic kinetic plot evaluated for the GeTe4 glass. Theoretical applicability of the JMA model as suggested by Málek [13] is displayed. The solid curve guides eyes in direction of the increasing particle size fraction of the GeTe4 glass
In the second step of kinetic analysis, the appropriate kinetic model is determined. One of the convenient ways is based on the values of α corresponding to the maxima of the z(α) and y(α) functions, α max,z and α max,y, respectively. In Fig. 3c, d, the values of α max,z and α max,y are plotted in dependence on particle size and against each other, respectively. In addition, the theoretical applicability of the JMA model is also suggested in the graphs—the “JMA fingerprint” is indicated by α max,z ≈ 0.632; however, it has recently been shown that this interval can be significantly extended without any loss of quality of the data description. As can be seen, the finest powder fraction (0–20 μm) and the coarse powders show a good correspondence with the suggested theoretical interval. This would normally mean that the crystallization process follows the JMA kinetics. Nevertheless, in the case of complex processes, it is always just the direct fit of the experimental data, which can confirm the validity of the JMA model (either the influence of a minor secondary process does not have to be recognized through the characteristic kinetic functions or two non-JMA processes with opposite asymmetry can compensate for each other so that the resulting signal has maximum at α max,z ≈ 0.632). On the other hand, the fine powder fractions (20–50 and 50–125 μm) and the bulk samples exhibit completely different behaviors from that expected for the JMA kinetics, where the kinetics of each fraction differs significantly from the other two.
As can be seen, even though relatively constant values of E and ΔH were obtained and kinetic plots (Fig. 3c, d) could be constructed, the basic single-peak kinetic analysis cannot provide a consistent satisfactory description of the observed complex kinetics. Reckless interpretation of these (at first sight) good kinetic data would provide completely erroneous conclusions (hence, the primary DSC data should always be provided together with any kinetic evaluations). Therefore, it is clear that in case of complex processes, the deconvolution procedures are mandatory in order to obtain correct and sound results.
Advanced kinetic analysis: deconvolution of kinetic data
In order to describe the complexity of the observed GeTe4 crystallization processes, the Fraser–Suzuki deconvolution procedure was applied to the data from the particle size fractions from 50–125 to 300–500 μm (those exhibiting multiple-peak behavior). The deconvolution was achieved without any restraining conditions for the optimization; no iterative fit-improving procedures needed to be used, which confirms the high accuracy and robustness of the deconvolution. An example of the deconvoluted complex crystallization peak is shown in Fig. 4—circles correspond to the experimental data, solid line represents the overall description (sum of the two sub-processes) and the two dashed lines correspond to the two underlying crystallization mechanisms. As can be seen, the crystallization process complexity can be described by two overlapping peaks of an opposite asymmetry (qualitatively similar deconvolution results were obtained for all complex DSC peaks). For descriptive purposes, the process with positive asymmetry will be labeled as autocatalytic (AC) and the negatively skewed peak will be labeled as JMA—justness of these denotations will be validated in the following paragraphs.

Example of the Fraser–Suzuki deconvolution procedure performed for the non-isothermal GeTe4 crystallization data (125–180 μm, q + = 1 °C min−1). Dashed lines correspond to the deconvoluted crystallization peaks; solid line represents the overall fit of the experimental data (circles)
In Fig. 5, the crystallization enthalpy ΔH and apparent activation energy E are displayed for the deconvoluted data (averaged over all applied heating rates for each particle size fraction). The gray circles then correspond to the data for the two finest powder fractions, where a single-peak behavior was observed—the data are shown for comparison. The crystallization enthalpy (magnitude) of the AC process decreases with d aver, while the enthalpy of the JMA process shows an opposite trend. This is in agreement with the recently found concept of competing CNT-based JMA crystal growth and autocatalytic crystallization from the mechanically induced heterogeneities (CNT stands for classical nucleation theory). This concept was confirmed for a number of chalcogenide glasses [21–26], where the two above-described processes occur when different powder fractions are studied. In accordance with this concept, the fraction of glassy matrix being crystallized via the CNT-based crystal growth mechanism increases with the portion of the “undamaged” bulk/surface material, where the formation of CNT nuclei can occur. Correspondingly, the autocatalytic crystallization from growth centers activated by grinding or milling procedures increases with the decreasing particle size (finer powders have much higher portion of their grains’ volume filled with mechanical defects and heterogeneities that can serve as crystallization centers).

a Crystallization enthalpies evaluated for the GeTe4 deconvoluted data and plotted in dependence on d aver. White and black symbols correspond to the deconvoluted AC and JMA peaks, respectively. The gray symbols show the overall ΔH values for the 0–20 and 20–50 μm powder fractions, for which the deconvolution was not performed (single-peak behavior occurred). b Activation energies evaluated for the deconvoluted data by the Kissinger (circles) and KAS (stars) methods and plotted in dependence on d aver. White and black symbols correspond to the deconvoluted AC and JMA peaks, respectively. The gray symbols show overall E values for the 0–20 and 20–50 μm powder fractions
The apparent activation energies of the deconvoluted processes plotted in Fig. 5b in dependence on d aver show that both AC and JMA mechanisms have relatively similar E values (confirmed for both original Kissinger and modified KAS methods). In agreement with our previous studies on other chalcogenide glasses [21–26], the activation energy for crystallization from mechanically induced heterogeneities is slightly higher than that found for the CNT-based crystal growth. Interestingly, in case of the 50–125 μm fraction, where the first notable occurrence of the two competing processes was observed, the E values of the two processes are reversed. Similar behavior was already observed in the case of As2Se3 glass [25, 27], where the CNT-based crystallization was partially suppressed by the presence of a high number of defects (hence higher E); the autocatalytic crystallization may then be expedited by the additional presence of newly formed crystallization centers originating from the stresses arising at the crystalline–amorphous phase interface.
Determination of the appropriate kinetic models for the deconvoluted processes is shown in Fig. 6. The first graph displays the α max,z and α max,y maxima for the particular studied particle size fractions. Based on the α max,z values and their comparison with the α max,z ≈ 0.632 JMA fingerprint value, the foregone denotation of the two types of deconvoluted processes (AC and JMA) is justified. In Fig. 6b, c the kinetic parameters are evaluated for the two respective applied kinetic models. The autocatalytic model was used also for description of the finest particle size fractions, even though the 0–20 μm fraction could also be described by the JMA model (the value of its kinetic parameter m was, however, unrealistically high, suggesting the JMA applicability being only apparent). Finally, the JMA sub-processes were found to have m = 2, indicating that growth of planar two-dimensional crystallites takes place in the GeTe4 powders.

a Particle size dependence of the characteristic kinetic functions maxima α max,z and α max,y evaluated for the GeTe4 data. White and black symbols correspond to the deconvoluted AC and JMA peaks, respectively. The gray symbols then show overall E values for the 0–20 and 20–50 μm powder fractions, for which the deconvolution was not performed (single-peak behavior occurred). Dashed line corresponds to the theoretical “fingerprint” α max,z = 0.632 value characteristic for the JMA model. b Kinetic parameters of the AC processes plotted in dependence on d aver. White symbols correspond to the deconvoluted AC peaks. The gray symbols show overall M and N values for the 0–20 and 20–50 μm powder fractions. c Kinetic parameter m evaluated for the deconvoluted JMA processes and plotted in dependence on d aver. Circles and stars correspond to the evaluations according to Málek [13] and Šesták [7], respectively
If we want to compare the present results with literature data, there is to the authors’ knowledge only one detailed kinetic study [28] dealing with non-isothermal crystallization kinetics of GeTe4. The reported particle size of the studied powder was ~50 µm, and the cooling during glass formation was, however, slightly more rapid than in the case of the present glass. This may be the reason for the lower apparent activation energy reported in [28]: E = 184 kJ mol−1, where the decrease of E would be associated with crystal growth-enhancing strains and defects originating from the glass forming. Regarding the model-based kinetics, the shape of the crystallization peaks shown in [28] is very similar to that of the presently reported data for the corresponding particle size fraction—exhibiting a positive asymmetry. Therefore, it is surprising that the authors [28] have assigned a two-dimensional growth to their data based on the double logarithm plot [7] because this crystallization mechanism is strictly associated with the JMA kinetics (which is inconsistent with the data). Based on this finding, one can derive that the double logarithm function is markedly insensitive to the selection of the actual model, which needs to be done outside the double logarithm-based evaluation. Only in case when the JMA kinetics is confirmed independently, the double logarithm function can be used to determine the kinetic parameter m JMA.
Structural interpretation of kinetic parameters
Identification of the crystallization mechanisms involved and their association with the formation of corresponding crystalline phases is the main task of crystallization kinetic studies. In the present section, the crystallization mechanisms found previously for the GeTe4 glass will be interpreted with regard to the structural information obtained from XRD and infrared microscopy.
In order to explain the crystallization behavior of GeTe4 powders, we will first look at crystal growth in the bulk material. The DSC bulk crystallization peaks were markedly different from the behavior of all the powder fractions (see Fig. 1). The slowly rising onset edges and sharply falling endset edges of the peaks are characteristic for the reaction order (F) and power law (P) models [29]. Detailed model-fitting analysis of the bulk DSC peaks has shown that the zero-order model (F0) provides the most appropriate f(α) function for their description:
Relevance of the F0 model was further tested by the application of the power law equation with the variable exponent n: f(α) = α n. Even in this case, the best fit indicated n = 0.051 ± 0.063. The congruity of the F0 model indicates that the amorphous-to-crystalline phase transformation is independent of the degree of conversion α and the rate of the transformation is equal to the rate constant given by the first term in Eq. (1). These conditions are typical for the true surface crystallization, where both nucleation and growth are catalyzed by the surface interface and defects present at this interface. In such case, the material is consumed at a rate of the progressing advancement of the amorphous/crystalline interface.
The validity of the above-suggested model for crystallization of bulk GeTe4 was further confirmed by infrared microscopy. It was found that the crystallization indeed starts from the surface of the sample, where a very fine crystalline microstructure is formed—see Fig. 7a (surface of the sample; view from above). At a further increase of temperature, the surface layer of fine crystallites slowly thickens and grows inwards until the whole sample undergoes the amorphous-to-crystalline transformation. In Fig. 7b, this layer of crystallites is displayed in a sample cross section—as can be seen, apart from occasional tiny glass defects, the crystallization proceeds strictly from the sample surface.

Micrographs obtained by means of infrared microscopy for the GeTe4 samples. a Surface layer of fine crystallites (view from above). b Cross-sectional view of a partially crystallized plate-like bulk sample—crystalline surface layer encloses the (yet) amorphous core. c Crystallite formed during slow cooling of GeTe4 melt. The left-side circular patterns indicate B-type spherulitic growth of a Te crystal
Comparing the crystallization behavior of the bulk and powders, it is clear that the crystal growth mechanism identified in the case of the bulk samples (F0) does not occur in the case of powder fractions. If the only difference between bulk and powders was the decreasing volume/surface ratio, the F0 crystallization mechanism would still show at least for coarse particle size fractions. The complete absence of this mechanism indicates that it is the mechanically induced defects and heterogeneities that take control of the nucleation and crystal growth in GeTe4 powders. The presence of the two distinct powder crystallization mechanisms (AC and JMA) and dependence of their relative manifestation on d aver further suggests that the two structurally different regions present in each glassy grain (surface layers and inner volume) play a role as well. Based on the findings obtained for the bulk GeTe4 sample, the explanation for powder crystallization can be derived: competitive crystal growth processes originating from the mechanically induced defects, where the heavily defective surface layer structures support the fast autocatalytic (AC) formation of a large amount of fine crystallites. On the other hand, the defects trenching deep within the grain volume (surrounded by undamaged glassy material) seem to surrogate/precipitate formation of CNT nuclei, which leads to the JMA(2) crystallization as a consequence.
Compositionally speaking, crystallization in GeTe4 glass is associated with the tellurium precipitation [30], followed by formation of the Te, GeTe and GeTe4 phases [28, 30, 31]. The occurrence of these phases apparently depends on a number of factors, the most important probably being amount and character of nuclei in the glassy material (where the nuclei may origin either from the glass-forming procedure or they may be associated with the sample processing), temperature regime (isothermal or non-isothermal crystallization), heating rates and annealing temperatures. Nevertheless, for the combinations of experimental conditions used within the framework of the current article, the formed crystalline phases were similar, as confirmed by XRD (see Experimental part). Hence, both the JMA and AC processes seem to be associated with the formation of compositionally similar crystallites—the primary growth of Te was followed by second-stage crystallization of GeTe. This may indicate a dominant influence of the first mentioned factor (density and character of nuclei in the material). Further confirmation of the above-stated finding can be deduced also from Fig. 7c, where the crystallite formed during the insufficiently fast melt quench is shown. The left part of the crystallite shows a well-formed spherulite, very similar to those growing in Se and Se–Te matrices [32, 33]. The presence of contrast rings indicates that a low-temperature “mode B” type of spherulite was formed [21, 32, 33]. As can be seen from the comparison of Fig. 7a, c (in both cases bulk material crystallized at a roughly similar temperature), the nucleation conditions and resulting character of the nuclei appear to strongly determine the final appearance of the formed crystallites.
Conclusions
Crystallization behavior of GeTe4 chalcogenide glass was studied by means of DSC, XRD and infrared microscopy. Deconvoluted non-isothermal DSC data obtained for wide range of particle size fractions were described in terms of the JMA and AC kinetic models. In addition, the zero-order kinetics was confirmed for the bulk samples. The large variety of observed kinetic mechanisms was explained on the basis of mechanically induced defects and heterogeneities, which accelerate the primary nucleation process. The marked importance of the presence of a defective structure on the initiation of crystal growth therefore explains the remarkable (considering the matrix being fully telluride) stability of the GeTe4 glass.
The XRD data confirmed that identical crystalline phases form regardless of the applied experimental conditions—the precipitation of Te is followed by second-stage GeTe crystal growth. This indicates the dominant influence of the nucleation processes on the consequent crystallization kinetics. Direct microscopic observations confirm the previous findings. In addition, type B tellurium spherulites were found to be formed under specific conditions, confirming the resemblance of Se/Te crystal morphologies.
References
Bureau B, Boussard-Plédel C, Lucas P, Zhang X, Lucas J. Forming glasses from Se and Te. Molecules. 2009;14:4337–50.
Sanghera JS, Aggarwal ID. Active and passive chalcogenide glass optical fibers for IR applications: a review. J Non-Cryst Solids. 1999;257:6–16.
Zakery A, Elliott SR. Optical properties and applications of chalcogendie glasses: a review. J Non-Cryst Solids. 2003;330:1–12.
Wilhelm AA, Boussard-Plédel C, Coulombier Q, Lucas J, Bureau B, Lucas P. Development of far-infrared-transmitting Te based glasses suitable for carbon dioxide detection and space optics. Adv Mater. 2007;19:3796–800.
Cui S, Chahal R, Boussard-Plédel C, Nazabal V, Doualan JL, Troles J, Lucas J, Bureau B. From selenium- to tellurium-based glass optical fibers for infrared spectroscopies. Molecules. 2013;18:5373–88.
Danto S, Houizot P, Boussard-Pledel C, Zhang XH, Smektala F, Lucas J. A family of far-infrared-transmitting Glasses in the Ga–Ge–Te system for space applications. Adv Funct Mater. 2006;16:1847–52.
Šesták J. Thermophysical properties of solids, their measurements and theoretical analysis. Amsterdam: Elsevier; 1984.
Kissinger HE. Reaction kinetics in differential thermal analysis. Anal Chem. 1957;29:1702–6.
Friedman HL. Kinetics of thermal degradation of char-forming plastics from thermogravimetry. Application to a phenolic plastic. New York: Wiley Subscription Services; 1964.
Starink MJ. The determination of activation energy from linear heating rate experiments: a comparison of the accuracy of isoconversion methods. Thermochim Acta. 2003;404:163–76.
Svoboda R, Málek J. Is the original Kissinger equation obsolete today? J Therm Anal Calorim. 2014;115:1961–7.
Málek J. Kinetic analysis of crystallization processes in amorphous materials. Thermochim Acta. 2000;355:239–53.
Málek J. The kinetic analysis of non-isothermal data. Thermochim Acta. 1992;200:257–69.
Avrami M. Kinetics of phase change I—general theory. J Chem Phys. 1939;7:1103–12.
Avrami M. Kinetics of phase change. II—transformation–time relations for random distribution of nuclei. J Chem Phys. 1940;7:212–24.
Avrami M. Granulation, phase change, and microstructure—kinetics of phase change. III. J Chem Phys. 1941;7:177–84.
Johnson WA, Mehl KF. Reaction kinetics in processes of nucleation and growth. Trans Am Inst Min (Metall) Eng. 1939;135:416–42.
Svoboda R, Málek J. Applicability of Fraser–Suzuki function in kinetic analysis of complex processes. J Therm Anal Calorim. 2013;111:1045–56.
Svoboda R, Málek J. Interpretation of crystallization kinetics results provided by DSC. Thermochim Acta. 2011;526:237–51.
Svoboda R, Málek J. Particle size influence on crystallization behavior of Ge2Sb2Se5 glass. J Non-Cryst Solids. 2012;358:276–84.
Svoboda R, Málek J. Thermal behavior in Se–Te chalcogenide system: interplay of thermodynamics and kinetics. J Chem Phys. 2014;141:224507.
Svoboda R, Málek J. Thermal behavior of Se-rich Ge2Sb2Se(5−y)Te y chalcogenide system. J Alloys Compd. 2015;81:101–8.
Svoboda R, Gutwirth J, Málek J, Wágner T. Crystallization kinetics of Se–Te thin films. Thin Solid Films. 2014;571:121–6.
Svoboda R, Brandová D, Málek J. Crystallization behavior of GeSb2Se4 chalcogenide glass. J Non-Cryst Solids. 2014;388:46–54.
Svoboda R, Málek J. Non-isothermal crystallization kinetics of As2Se3 glass studied by DSC. Thermochim Acta. 2014;579:56–63.
Svoboda R, Přikryl J, Barták J, Vlček M, Málek J. Crystallization behavior of Ge17Sb23Se60 thin films. Philos Mag. 2014;94:1301–10.
Svoboda R, Málek J. Nucleation in As2Se3 glass studied by DSC. Thermochim Acta. 2014;593:16–21.
Abu-Sehly AA. Study of the kinetics of non-isothermal crystallization in Ge20Te80 chalcogenide glass. Phys B. 2003;325:372–9.
Khawam A, Flanagan DR. Solid-state kinetic models: basics and mathematical fundamentals. J Phys Chem B. 2006;110:17315–28.
Quinn RK, Johnson RT. Thermally-induced structural and electrical effects in Ge–Te-based amorphous alloys. J Non-Cryst Solids. 1973;12:213–31.
Wang G, Nie Q, Wang X, Dai S, Xu T, Shen X, Zhang X. Composition dependence of optical band gap of the Se–Ge–Te far infrared transmitting glasses. Phys B. 2010;405:4424–8.
Ryschenkow G, Faivre G. Bulk crystallization of liquid selenium. J Cryst Growth. 1988;87:221–35.
Bisault J, Ryschenkow G, Faivre G. Spherulitic branching in the crystallization of liquid selenium. J Cryst Growth. 1991;110:889–909.
Acknowledgements
This work has been supported by the Czech Science Foundation under Project No. P106/11/1152.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Svoboda, R., Brandová, D. & Málek, J. Non-isothermal crystallization kinetics of GeTe4 infrared glass. J Therm Anal Calorim 123, 195–204 (2016). https://doi.org/10.1007/s10973-015-4937-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-015-4937-x