
Abstract
This work describes how the silica/alumina ratio (SAR) affects the framework, texture, chemical composition, and acidity of SAPO-34 molecular sieves synthesized via hydrothermal crystallization using morpholine (MOR) or triethylamine (TEA) as structure-directing agents (SDA’s). The solids were characterized by X-ray diffraction (XRD), scanning electron microscopy (SEM), N2 adsorption-desorption isotherms, thermogravimetric analysis (TGA), Fourier-transform infrared spectroscopy (FT-IR), X-ray fluorescence spectrometry (XRF), silicon-29 MAS NMR spectroscopy (29Si MAS NMR), aluminum-27 MAS NMR spectroscopy (27Al MAS NMR), and temperature-programmed desorption of ammonia (NH3-TPD). The solids synthesized in this work had crystalline phases, vibrational modes, and morphology characteristics of silicoaluminophosphate (SAPO) materials. Pure SAPO-34 was obtained only with morpholine, while SAPO-18/34 was crystallized with TEA. The SAPO-34 synthesized with MOR had the higher relative crystallinity and incorporation of Si into the framework. The SAPO-18/34 prepared with TEA had the larger surface areas. The solids prepared with SAR 0.3 had only Si(4Al) coordination, independently of the SDA used. The SAPO-34 synthesized with MOR and SAR 0.6 had the highest total acidity (9.75 μmol NH3 m−2).

SAPO molecular sieves were synthesized hydrothermally with different structure-directing agents and silica/alumina ratios. The effects of both variables on the physical and chemical properties of the samples obtained were evaluated. Pure SAPO-34 was obtained with morpholine, while SAPO-18/34 was crystallized with TEA. The SP34T-0.6 sample prepared with triethylamine and SAR 0.6 showed promising characteristics for use as catalyst in MTO reactions: AEI/CHA intergrowths, the lowest acidity, the smallest crystallite size, and a large BET surface area.
Highlights
-
Simultaneous effects of SAR and MOR or TEA on the SAPO-34 properties were studied.
-
SAPO-34 synthesized with morpholine (MOR) had the higher relative crystallinity.
-
AEI/CHA intergrowths were synthesized with triethylamine (TEA) and had the larger surface areas.
-
SAPO prepared with a silica/alumina ratio (SAR) of 0.3 had only the Si(4Al).
-
The SAPO-18/34 synthesized with TEA and SAR 0.6 had the lowest total acidity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Zeolites are used in processes such as energy storage, water treatment, environmental control, separation, and heterogeneous catalysis owing to their organized microporous structure, which allows the selective separation and conversion of molecules according to size and chemical structure [1]. In particular, the thermal stability, molecular selectivity, and surface acidity (mild acidity) of the SAPO-34 are responsible for its application in catalysis, being an especially useful catalyst to produce light olefins [2,3,4,5,6,7,8]. According to Akhgar et al. [9] and Bahrami et al. [10], SAPO-34 is one of the catalysts with better performance in the methanol-to-olefin (MTO) conversion processes. It is worth mentioning that the SAPO-18/34 intergrowth materials are also promising catalysts for these processes, as reported by Zhao et al. [6].
The SAPO-34 molecular sieve is isomorphic to the chabazite zeolite (CHA) with pores 0.43 × 0.43 nm in size [11]. The tridimensional cages of SAPO-34 are 0.75 × 0.82 nm in size [2] and are connected by six-member double rings [10]. Molecules such as primary alcohols, linear paraffins, and olefins can flow through pore sizes with these dimensions, while branched isomers and aromatic compounds, which can be formed in the cage, cannot flow through the SAPO-34 pores, thus narrowing the product distribution [2, 4].
SAPO-34 was first synthesized in 1984 by researchers at Union Carbide Corporation [11] and it is usually prepared via hydrothermal crystallization of silicoaluminophosphate gels using an organic compound, such as an amine, as a structure-directing agent (SDA). The SDA determines the structure of the solid by filling its empty spaces and by producing a charge compensation effect that is necessary after substitution of the Si in the framework by Al atoms [8, 12]. Amines as tetraethylammonium hydroxide (TEAOH), diethylamine (DEA), triethylamine (TEA), morpholine (MOR), piperidine, and combinations of these compounds, such as TEAOH/DEA, have been used as SDA in SAPO-34 synthesis [1,2,3, 5,6,7,8,9,10, 12]. Furthermore, other SDA molecules like 4-4′-trimethylene-dipiperidine (TMDP) can be used to synthesize silicoaluminophosphates such as HSAPO-1 and SAPO-L [13].
Thus, the relative crystallinity, crystallite size, texture, and morphology of the SAPO-34 depend on the SDA used in the synthesis, since it acts as a template during incorporation of Si into the framework [8, 9, 14,15,16]. In addition, the chemical composition of the unit cell and the acidity of the SAPO molecular sieves also depend on the number and distribution of Si atoms in the framework and hence on the Si/Al ratio (SAR) used in the starting gel, the SDA, and synthesis parameters such as temperature and pH [4, 12, 14, 17]. Each unit cell in the SAPO-34 molecular sieve has three cavities composed of 36T atoms (T = Si or Al or P) [18]. Also, different Si(nAl) coordination (n from 0 to 4) results in different acidic strengths, depending on the electronegativity of each tetrahedron (TO4, T = Si or Al or P) set around a central Si atom. The acidity decreases in the following order: Si(1Al) > Si(2Al) > Si(3Al) > Si(4Al) > Si(0Al) [12].
Morpholine (MOR) and triethylamine (TEA) are useful structure-directing agents for the synthesis of SAPO molecular sieves [3, 9, 10, 16, 18, 19]. However, it is necessary to expand our knowledge about the influence of these templates, with different SAR’s, on the formation and properties of SAPO-34 crystallites. Thus, the effect of MOR and TEA on the structure of SAPO-34 must be studied for different Si concentrations in the starting gel and synthesis methodologies. In this work, we addressed the effect of SAR and SDA on the structural properties of SAPO-34 molecular sieves synthesized via hydrothermal crystallization of gels with different molar compositions.
2 Experimental
2.1 Solids preparation
The SAPO-34 molecular sieves were synthesized via hydrothermal crystallization of gels with molar composition given by the formula 3SDA:xSiO2:Al2O3:P2O5:60H2O, where x is the SiO2:Al2O3 molar ratio (0.3, 0.6, or 0.9). Morpholine (99.0 %wt., Merck) and triethylamine (99.98 %wt., Merck) were used as SDA. Pseudobohemite (99.2 %wt., Al2O3, Grace), orthophosphoric acid (85 %wt., Merck), and silica sol (99.8 %wt., SiO2 Aerosil® 200, Degussa-Hüls) were used as the sources of aluminum, phosphorous, and silicon, respectively. The solids synthesized with morpholine and triethylamine were named SP34M and SP34T, respectively, followed by the SAR used in the starting gel (e.g. SP34M-0.3, SP34T-0.3). Initially, a solution containing orthophosphoric acid, deionized water, and pseudobohemite was stirred at 20 °C for 6 h. In a different flask, a solution of silica, SDA, and deionized water was stirred at room temperature for 1 h. Then both solutions were mixed in a 250 ml round flask. The resulting solution was stirred for 24 h at 27 °C. The aged gel was subsequently transferred to a Teflon vessel enclosed in a stainless-steel jacket and heated in a muffle oven at 200 °C for 48 h. Then the vessel was cooled to room temperature and the solution was filtered, washed with deionized water, and dried at 110 °C for 12 h. Finally, the solids were calcined at 600 °C for 6 h at an airflow rate of 30 ml min−1.
2.2 Solids characterization
The crystalline structure of the samples was determined by X-ray diffraction in a Philips Analytical X-ray Diffractometer (X´Pert-MPD, Netherlands) with CuKɑ radiation (λ = 0.154056 nm), operated at 40 kV, 40 mA, and a scanning rate of 0.05° min−1. The crystallite size (d) was calculated from the XRD spectra using Scherrer’s equation (Eq. 1) [9].
where λ is the radiation wavelength, B(2θ) is the peak width at full width half maximum (FWHM) (2θ = 20.5°), and θ is the angle in radians. Gaussian functions were used to adjust diffractogram peaks and estimate the crystallite sizes. The relative crystallinity of the solids was calculated from the main peak intensities. The morphology of the samples was observed by scanning electron microscopy using a scanning electron microscope LEO Electron Microscopy Oxford (440i, England). The textural properties were obtained by N2 adsorption-desorption isotherms at 77 K in a Micromeritics ASAP (2020, USA). The surface areas and pore volumes were calculated by the Brunauer–Emmett–Teller (BET) and t-plot methods, respectively. The weight loss was measured by thermogravimetric analysis (TGA) performed in a Shimadzu TGA (TGA-50M, Japan) between 25 and 1000 °C at 10 °C min−1 and an airflow of 25 mL min−1. The chemical bonds and vibrational modes were observed by Fourier-transform infrared spectroscopy (FT-IR) carried out in a Thermo Scientific Nicolet (6700, USA) using the KBr method from 400 to 4000 cm−1 with a resolution of 4 cm−1 and 32 scans. The chemical composition of the solids was measured by X-ray fluorescence analysis in a Shimadzu (XRF1800, Japan) spectrometer, using a rhodium source and operating at 40 kV, 95 mA, a rotation of 8° min−1, and a step size of 0.1°. The Si and Al coordinations were measured by silicon-29 magic angle spinning NMR spectroscopy (29Si MAS NMR) and by 27Al MAS NMR, respectively, carried out at room temperature in a Bruker spectrometer (AC 300/P). For the 29Si MAS NMR analysis, the equipment was operated at 79.5 MHz with an acquisition time of 0.0409600 s, a contact time of 5 ms, a delay time of 1 s, and a spectral window of 315 ppm. The 27Al MAS NMR spectra were obtained at 105 MHz, an acquisition time of 0.0327680 s, a contact time of 1 ms, a delay time of 2 s, and a spectral window of 2400 ppm. The surface concentration of acid sites of the solids was determined by temperature-programmed desorption of ammonia (NH3-TPD) using a Micromeritics Autochem II (2920, USA) with a coupled thermal conductivity detector (TCD). Approximately 0.2 g of sample were preheated at 500 °C for 1 h in helium and then cooled to 100 °C for saturation with flowing 10 %NH3/He at 15 mL min−1 for 30 min. Helium was used to purge the sample for 30 min. Then the sample was heated to 700 °C at 10 °C min−1 for desorption of the adsorbed ammonia.
3 Results and discussion
3.1 Structural, morphological, and textural properties of the SAPO-34
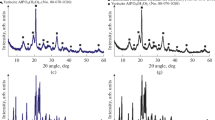
The X-ray diffraction patterns of the solids synthesized with MOR (Fig. 1) exhibit all the main peaks seen in SAPO-34 molecular sieves in accordance with the International Zeolite Association [20] standard, suggesting the formation of SAPO-34 crystals with no or only small amounts of contaminant phases. For the samples prepared with TEA, the XRD patterns are significantly different.

XRD patterns of solids synthesized
The relative crystallinity and the mean crystallite size (Table 1) of the samples synthesized with MOR increased with increasing SAR, in agreement with the work of Prakash and Unnikrishnan [18], suggesting that SiO2 concentration strongly affects crystal size when MOR is used as SDA. The reverse trend was observed for the samples synthesized with TEA. However, the crystallite size of these solids does not change much with SAR and is in the range of 35–56 nm, which corroborates the nano-structure framework of the samples in agreement with results obtained by Akhgar et al. [9] when studying hierarchical SAPO-34 with SAR 0.6. According to Liu et al. [16] and Xu et al. [19], the low relative crystallinity of the samples synthesized with TEA is probably due to distortions induced by the formation of a eutectic mixture between the crystalline phases of the SAPO-34 (CHA structure) and SAPO-18 (AEI structure) molecular sieves. These distortions produce high intensity of the peaks at low angles and low intensity of the peaks at high angles, as shown by Liu et al. [16]. Therefore, because of this structural disorder of the samples prepared with TEA and the effect of superimposition on weak reflections, the relative crystallinity of these samples cannot be reliable.
Compared with the XRD patterns found in the literature [21], the patterns of samples prepared with TEA (Fig. 1) indicate AEI/CHA intergrowths with an AEI stacking probability of 40–50%. The CHA and AEI phases have the same basic structure composed of six-membered double rings, forming different openings and inducing the crystallization of both phases, which are formed with different ligands of the oxygen bridges. Comparing CHA with AEI, the AEI cage of 1.16 nm is slightly wider than that of CHA (0.94 nm), but the difference in cage shape does not change the pore diameter, that is the same of 0.43 nm in SAPO-34 and SAPO-18 [6, 22], and was reported as 0.47 for a SAPO-18/34 synthesized by Zhao et al. [23]. Therefore, the main effect of TEA as SDA is crystallization of the AEI/CHA intergrowths, while MOR favors crystallization of the CHA-type molecular sieve for all the SAR used, in agreement with the results of the Liu et al. [16] study using samples prepared with SAR 0.6.
The micrographs of the synthesized solids (Fig. 2) show cubic morphologies characteristic of the SAPO-34 and SAPO-18/34 molecular sieves [5, 6, 8, 9, 24]. The increase in SAR from 0.3 to 0.9 increased the formation of smaller crystallites, amorphous structures, and agglomerate crystallites, irrespective of the SDA used. The solids prepared with MOR and SAR 0.6 and 0.9 had larger crystallite sizes than those prepared with TEA, in agreement with the XRD analysis (Table 1) and with the work of Liu et al. [16]. According to Ye et al. [8], the nature of the template used in the synthesis process determines the morphology of final crystals due to different rates of crystal growth.

Micrographs of the synthesized solids obtained by SEM
Figure 3 shows the N2 adsorption-desorption isotherms of the samples. The solids prepared with SAR 0.3 and 0.6, using MOR or TEA, showed type I isotherms, which are typical of microporous molecular sieves with small external surface areas [5, 6]. The solids synthesized with SAR 0.9 (SP34M-0.9 and SP34T-0.9) showed a mixture of type I and IV isotherms, indicating that the solids have microporous and mesoporous structures. Also, the type H1 hysteresis loop was observed, which corresponds to uniform pores with a narrow pore size distribution [25], in accordance with the X-ray diffraction patterns (Fig. 1). Thus, the increase in SiO2 content in the synthesis gel for SAR higher than 0.6 increased the fraction of mesopores in the solids, irrespective of the SDA used.

N2 adsorption (+)/desorption (ο) of the solids prepared with MOR (a) and TEA (b)
The SAR and SDA strongly affect the textural properties of the solids (Table 2). The BET surface area of the SAPO-34 synthesized with MOR and SAR 0.3 and 0.6 was about 415 m2 g−1, whereas the surface area of the sample prepared with SAR 0.9 was about 464 m2 g−1, an increase of 12%. The samples synthesized with TEA had larger surfaces areas than those prepared with MOR for all SAR used in this work, which can be related to the structure of these samples, characterized by the AEI/CHA intergrowths. Zhao et al. [6] reported BET surface areas for AEI/CHA intergrowth zeolites of 530.4–579.5 m2 g−1, in agreement with the values observed for the SP34T samples (Table 2). Also, the SAPO-18/34 prepared with TEA showed a decrease in surface area as the SAR increased. According to Akhgar et al. [9] and Bahrami et al. [10], the micropore surface area (Table 2) decreases with decreasing crystallinity (Fig. 1), explaining the tendency of micropore surface areas obtained, except in samples SP34M-0.3 and SP34M-0.6. The external surface area of all samples increased as the Si content increased.
The thermogravimetric analysis of the dried samples showed significant mass losses in three temperature ranges (Table 3). The first was observed from 25 up to 300 °C and corresponds to the desorption of water from the pores of the molecular sieves. The second was observed between 300 and 600 °C and was assigned to the thermal decomposition of the SDA and organic residues. The third peak, observed above 600 °C, is attributed to the removal of organic residues from the deeper cavities of the solids during calcination in an oxygen-rich atmosphere [15, 16]. Different mass losses were obtained for each solid in the three temperature ranges. The samples synthesized with MOR showed the higher total mass losses, with a mean of 20.5%. For the samples synthesized with TEA, the lower mass losses were observed at the third temperature range, irrespective of the SAR used in the starting gel, which agrees with their smaller crystals identified by XRD (Table 1). These results suggest that different solids were obtained and that MOR produces SAPO-34 with deeper cavities than SAPO-18/34 prepared with TEA, which could promote the incorporation of Si into the framework. Hence, the type of SDA strongly affects the distribution of organic and water molecules inside the pores during the formation of the molecular sieves, as also shown by the results on crystallinity (Fig. 1) and surface area (Table 2).
3.2 Chemical properties of the SAPO-34
The FT-IR spectra of the dry samples are shown in Fig. 4. The infrared bands observed for zeolites (1300–400 cm−1) are commonly used to analyze the inorganic structure of the molecular sieves. The bands observed at 424, 480, 530, 570, 635, 730, 1100, and 1215 cm−1 in Fig. 4b correspond to symmetric and asymmetric stretches of chemical bonds between P, O, Si, and Al atoms as well as six- or eight-member double rings, typical of the CHA and AEI phases. This result suggests the formation of the SAPO-34 and SAPO-18/34 structures [5, 9, 17, 26, 27] and agrees with the XRD and SEM results (Figs. 1 and 2, respectively). In Fig. 4a, the band at 1645 cm−1 was assigned to the H–O–H bond of water weakly bonded to the SDA (physical adsorption) in the framework in accordance with the work of Wang et al. [5] and Akhgar et al. [9]. The bands at 3590 and 3625 cm−1 are assigned to the stretching of Brønsted acid sites attributed to the Si–OH–Al groups [5, 9, 26]. The peaks at 868 and 816 cm−1 correspond to protonated morpholine molecules in the SAPO-34 structure [18, 27]. The bands at 1400, 1540, and 3000 cm−1 correspond to C-H bonds and are assigned to SDA molecules inside the pores [3, 13, 28].

FT-IR spectra in the regions (a) 1300–4000 cm−1 and (b) 400–1300 cm−1 of the solids synthesized
The molar fraction of Si in the solids is higher than that in the precursor gel (Table 4), suggesting that during crystallization the incorporation of Si atoms into the SAPO framework is larger than that of P and Al atoms; and, according to the elemental compositions, there is more incorporation of P atoms than Al in all samples. According to Tan et al. [17], during crystallization Al, P, and Si atoms are initially incorporated into the framework, but after 2.5 h the Al and P atoms are substituted by Si atoms, increasing the relative content of Si. Also, the solids synthesized with MOR have a higher molar fraction of Si than those prepared with TEA, in accordance with previous work [16], indicating that Si incorporation may be related to the larger crystallite size (Table 1) and smaller surface areas (Table 2).
Table 5 shows the chemical composition of the unit cell of the solids calculated from XRF (Table 4) and TGA (Table 3) results. A significant variation in the number of SDA molecules per cage in the composition of the unit cells of samples prepared with MOR and TEA is observed. The SAPO-34 prepared with MOR showed the higher number of SDA molecules per cage. However, the Si/Al ratios in the unit cell were similar in all the samples prepared with the same SAR and with MOR or TEA as SDA, suggesting that the Si/Al ratio in the unit cell only depends on the SAR in the starting gel, irrespective of the SDA used. In addition, the number of MOR molecules per cage for SP34M-0.3 and SP34M-0.9 is higher than the theoretical maximum of two [18]. This deviation of about 10% from the maximum reported was tolerable and was considered to be the inaccuracy of the method of analysis.
The 29Si MAS NMR spectra were used to determine the number of Al atoms bonded to one Si atom. Figure 5a shows the relative position of several Si(nAl) coordinations in which n is 0, 1, 2, 3, or 4, meaning that the SiO4 tetrahedron is bonded to n AlO4 tetrahedra [4]. The peak at −85.5 ppm is assigned to the Si(4Al) coordination, which results from the substitution of a phosphorous atom in the structure by a silicon atom during crystallization [8]. This mechanism is known as SM2 and occurs when Si concentration is low [19]. The peaks at −89.5, −99, −105, and −108 ppm are assigned to Si(3Al), Si(2Al), Si(1Al), and Si(0Al) coordinations in the structural network, respectively [8, 29], suggesting that the predominant substitution mechanism was SM3, which consists in the substitution of neighboring Al and P atoms by two Si atoms and occurs at high Si concentrations [29, 30]. Also, the signal of these peaks exhibits an upfield shift that increases as the number of AlO4 tetrahedra (n) bonded to a silicon atom decreases [4].

Si29 (a) and Al27 (b) MAS NMR spectra of the solids synthesized
The 27Al MAS NMR spectra (Fig. 5b) show peaks at 38 ppm assigned to the tetrahedral coordination of the Al, Al(4P), typical of the SAPO molecular sieves. The low-intensity peak at 15 ppm corresponds to the pentahedral coordination of the Al and secondary interactions of the aluminum structure with water or template. Peaks at −11 ppm correspond to the octahedral Al atoms [3, 18, 31]. A large number of octahedral Al species could be explained by the presence of entrapped template or water in the adjacent channels, forming Al(OP)4(H2O)2 octahedral coordination, for example [31]. Especially the presence of water can have a considerable effect on local symmetry around the Al atom [31].
Table 6 lists the quantification of the silicon atom coordination in the solids, calculated by fitting Gaussian curves and deconvolution of the NMR peaks (Fig. 5a). When the (P + Si)/Al ratio is equal to 1, Si incorporation into the AlPO structure takes place either in the crystallization step or by a SM2 substitution mechanism with the formation of the Si(4Al) coordination [19]. For (P + Si)/Al greater than 1, the Si atom is incorporated into the AlPO structure via substitution mechanism SM3 with formation of the Si(nAl) coordination with n varying from 3 to 0.
In the 29Si MAS NMR spectra, only the SP34M-0.3 and SP34T-0.3 solids show one single peak, located at −85.5 ppm, which corresponds to the Si(4Al) coordination. This single peak was observed even for (P + Si)/Al ratios greater than 1 (Table 4), which in principle, should induce formation of Si(nAl) with n from 3 to 0 (SM3 mechanism). The fact that only one peak is observed suggests that the SDA can be responsible for charge compensation in the SAPO structure, thus favoring substitution of P atoms by Si atoms in the crystallization step via an SM2 mechanism. The SP34M-0.6 and SP34M-0.9 solids have an additional peak at −99 ppm which is normally assigned to Si(2Al) coordination, indicating the existence of the SM2 and SM3 mechanisms, simultaneously. This is explained by the increase in the (P + Si)/Al ratio and the consequent decrease in the Si(4Al) coordination in the solids structure. This is also observed in the solids synthesized with TEA and SAR greater than 0.3. Also, the coordination number is greater than that obtained for the samples prepared with MOR. Xu et al. [19] reported the same behavior for the increase in SAR in molecular sieves prepared with TEA. The SP34T-0.9 sample is the only one that shows five types of Si coordination, which can be explained by its Si/(Al + P + Si) ratio (0.257) and (P + Si)/Al ratio (1.957) being higher than those of the other SAPO prepared in this work, as shown in Table 4. Moreover, except for SP34T-0.9, for all samples there is prevalence of the SM2 mechanism, indicating that P sites were preferentially get substituted by Si.
The molecular sieves synthesized with MOR and SAR higher than 0.3 had more Si(4Al) species than did those prepared with TEA, in good agreement with the work of Liu et al. [16]. For example, for solids prepared with MOR it was observed that relative crystallinity (Table 1) decreased as Si(4Al) coordination increased, whereas for solids prepared with TEA the opposite was observed. Studies have shown that Si islands with different coordinations can make it more difficult for crystallite to grow and affect molecular sieve crystallinity [32].
Figure 6 contains the NH3-TPD profiles for the solids prepared in this work. Two large peaks are observed for all samples, corresponding to weak and strong acid sites, in accordance with Wang et al. [5], Ye et al. [8], Akhgar et al. [9], and Zhao et al. [23]. The peak observed between 100 and 300 °C corresponds to weak Brønsted acid sites attributed to the surface hydroxyl groups (–OH), which are limited by defective sites such as P–OH, Si–OH, and Al–OH that are not completely connected to the tetrahedral AlO4 [1, 7, 9, 33]. The second peak, observed between 300 and 650 °C, corresponds to strong Brønsted acid sites attributed to hydroxyl group ligands such as –Si–OH–Al– [7,8,9]. These sites are produced as charge compensation caused during the substitution of P atoms by Si atoms in the structure of neutral AIPO molecular sieves [4, 8]. Moreover, these hydroxyl groups affect catalytic performance in the MTO process, since production of light olefins depends on the concentration of these acid sites [8,9,10].

Temperature-programmed desorption of ammonia of the solids synthesized
The symmetry of the peaks in the NH3-TPD profiles made it easy to quantify the density of weak, strong, and total acid sites (Table 7) via deconvolution and Gaussian fits [7, 27].
The lower total acid densities were seen in SP34T-0.6 and SP34M-0.9. The samples with SAR 0.3 and 0.6 prepared with MOR had higher total acid densities than those with corresponding SAR prepared with TEA. This can be explained by the larger number of molecules of SDA per cage (Table 5) and higher Si incorporation (Table 4) shown by the samples prepared with MOR than those shown by the samples prepared with TEA. According to Akhgar et al. [9], acidity is related to the Si incorporated into the crystalline framework. Therefore, these results suggest the important effect of the SDA on the acidity of the SAPO, which is directly related to the Si distribution in its framework.
The SAPO-34 synthesized with MOR and SAR 0.6 had the highest total density of acid sites, whereas the sample prepared with MOR and SAR 0.9 had a relatively low total density of acid sites in spite of the high Si incorporation (Table 4), the presence of Si(2Al) coordinations, and the location of more than 90% of the Si atoms in the Si(4Al) chemical environment (Table 6). These results suggest that not all the Si atoms incorporated into the SAPO-34 framework are bonded to acidic protons owing to the different coordination of the Si atoms, even when Si-OH-Al species were incorporated into the framework [2]. In addition, high Si concentrations in the synthesis gel result in a large number of SiO4 tetrahedra, which reduces the number of Si-OH-Al species and, consequently, the acid sites [14].
4 Conclusions
In this work, the effect of SAR on structural, textural, and chemical properties of SAPO-34 molecular sieves prepared with different structure-directing agents (MOR and TEA) was studied. Crystals, vibrational modes, microporous structure, and morphology typical of SAPO materials were confirmed by XRD, FT-IR, N2 physisorption, and SEM analysis. The pure SAPO-34 synthesized with MOR showed the higher relative crystallinity and Si incorporation into the framework, whereas solids synthesized with TEA showed AEI/CHA intergrowths for all the SAR used. The increase in SAR of the synthesis gel above 0.6 increased the fraction of mesopores in the solids, irrespective of the SDA used. The solids prepared with TEA showed the larger surface areas. The SAPO-18/34 prepared with TEA and SAR 0.9 showed the widest silicon environment distribution, whereas only the Si(4Al) coordination was obtained in the SAPO prepared with SAR 0.3, irrespective of the SDA used. The 27Al MAS NMR analysis demonstrated the existence of Al species in tetrahedral, pentahedral, and octahedral coordination. The increase in SAR of the synthesis gel produced changes in the acid site density of the SAPO. The use of MOR favored the formation of acid sites for samples with SAR up to 0.6. The SAPO-34 synthesized with MOR and SAR 0.6 showed the highest total acid density (9.75 μmol NH3 m−2).
SP34T-0.6 has the smallest crystallite size, one of the larger BET surface areas, and the lowest acidity of the samples. Thus, also considering the results of Zhao et al. [6], Akhgar et al.[9], and Aguayo et al. [22], it is expected that SAPO-18/34 synthesized with TEA and SAR 0.6 has the highest olefin yield and longest catalyst lifetime in the MTO process.
Data availability
The data that support the results of this study are available on request to the corresponding author, GPV.
References
Cui Y, Zhang Q, He J, Wang Y, Wei F (2013) Pore-structure-mediated hierarchical SAPO-34. Particuology, https://doi.org/10.1016/j.partic.2012.12.009
Wilson S, Barger P (1999) The characteristics of SAPO-34 which influence the conversion of methanol to light olefins. Microporous Mesoporous Mater, https://doi.org/10.1016/S1387-1811(98)00325-4
Gao B, Fan D, Sun L, An H, Fan F, Xu S, Tian P, Liu Z (2017) Insights into the aminothermal crystallization process of SAPO-34 and its comparison with hydrothermal system. Microporous Mesoporous Mater, https://doi.org/10.1016/j.micromeso.2017.04.035
Pinilla-Herrero I, Olsbye U, Márquez-Álvarez C, Sastre E (2017) Effect of framework topology of SAPO catalysts on selectivity and deactivation profile in the methanol-to-olefins reaction. J Catal, https://doi.org/10.1016/j.jcat.2017.05.008
Wang J, Yang M, Shang W, Su X, Hao Q, Chen H, Ma X (2017) Synthesis, characterization, and catalytic application of hierarchical SAPO-34 zeolite with three-dimensionally ordered mesoporous-imprinted structure. Microporous Mesoporous Mat, https://doi.org/10.1016/j.micromeso.2017.06.012
Zhao D, Zhang Y, Li Z, Wang Y, Yu J (2017) Synthesis of AEI/CHA intergrowth zeolites by dual templates and their catalytic performance for dimethyl ether to olefins. Chem Eng J, https://doi.org/10.1016/j.cej.2017.04.109
Izadbakhsh A, Farhadi F, Khorasheh F, Yan ZF (2009) Effect of heating profile on desorption curve in temperature programmed desorption analysis. J Porous Mater, https://doi.org/10.1007/s10934-008-9237-4
Ye L, Cao F, Ying W, Fang D, Sun Q (2011) Effect of different TEAOH/DEA combinations on SAPO-34’s synthesis and catalytic performance. J Porous Mater, https://doi.org/10.1007/s10934-010-9374-4
Akhgar S, Towfighi J, Hamidzadeh M (2020) A green and cost-effective surfactant-assisted synthesis of SAPO-34 using dual microporous templates with improved performance in MTO reaction. J Sol Gel Sci Technol, https://doi.org/10.1007/s10971-020-05308-w
Bahrami H, Darian JT, Sedighi M (2018) Simultaneous effects of water, TEAOH and morpholine on SAPO 34 synthesis and its performance in MTO process. Microporous Mesoporous Mat, https://doi.org/10.1016/j.micromeso.2017.11.011
Lok BM, Messina CA, Patton RL, Gajek RT, Cannan TR, Flanigen EM (1984) Silicoaluminophosphate molecular sieves. J Am Chem Soc, https://doi.org/10.1021/ja00332a063
Yu T, Fan D, Hao T, Wang J, Shen M, Li W (2014) The effect of various templates on the NH3-SCR activities over Cu/SAPO-34 catalysts. Chem Eng J, https://doi.org/10.1016/j.cej.2014.01.008
Sarkar K, Laha SC, Bhaumik A (2006) A new extra large pore organic-inorganic hybrid silicoaluminophosphate. J Mater Chem, https://doi.org/10.1039/B600989A
Vomscheid R, Briend M, Peltre MJ, Man PP, Barthomeuf D (1994) The role of the template in directing the Si distribution in SAPO zeolites. J Phys Chem, https://doi.org/10.1021/j100089a041
Álvaro-Muñoz T, Márquez-Álvarez C, Sastre E (2012) Use of different templates on SAPO-34 synthesis. Catal Today https://doi.org/10.1016/j.cattod.2011.07.038
Liu G, Tian P, Liu Z (2012) Synthesis of SAPO-34 molecular sieves templated with diethylamine and their properties compared with other templates. Chin J Catal, https://doi.org/10.1016/S1872-2067(10)60325-2
Tan J, Liu Z, Bao X, Liu X, Han X, He C, Zhai R (2002) Crystallization and Si incorporation mechanisms of SAPO-34. Microporous Mesoporous Mat, https://doi.org/10.1016/S1387-1811(02)00329-3
Prakash AM, Unnikrishnan S (1994) Synthesis of SAPO-34. J Chem Soc Faraday Trans, https://doi.org/10.1039/FT9949002291
Xu L, Du A, Wei Y, Wang Y, Yu Z, He Y, Zhang X (2008) Synthesis of SAPO-34 with only Si(4Al) species: effect of Si contents on Si incorporation mechanism and Si coordination environment of SAPO-34. Microporous Mesoporous Mat, https://doi.org/10.1016/j.micromeso.2008.02.001
IZA, International Zeolite Association (2020) Database of zeolite structures. http://www.iza-online.org. Accessed 08 Feb 2021
Lillerud KP, Akporiaye D (1994) Systematic relationships between the structures of CHA, AEI and KFI. Stud Surf Sci Catal, https://doi.org/10.1016/S0167-2991(08)64156-7
Aguayo AT, Gayubo AG, Vivanco R, Olazar M, Bilbao J (2005) Role of acidity and microporous structure in alternative catalysts for the transformation of methanol into olefins. Appl Catal A Gen, https://doi.org/10.1016/j.apcata.2005.01.006
Zhao D, Zhang Y, Li Z, Wang Y, Yu J (2017) Synthesis of SAPO-18/34 intergrowth zeolites and their enhanced stability for dimethyl ether to olefins. RSC Adv, https://doi.org/10.1039/c6ra25080g
Liu G, Tian P, Zhang Y, Li J, Xu L, Meng S, Liu Z (2008) Synthesis of SAPO-34 templated by diethylamine. Microporous Mesoporous Mat, https://doi.org/10.1016/j.micromeso.2008.01.030
Sing KSW, Everett DH, Haul RAW, Moscou L, Pierotti RA, Rouquérol J, Siemieniewska T (1985) Reporting physisorption data for gas/solid systems. Pure Appl Chem, https://doi.org/10.1351/pac198557040603
Breck DW (1974) Zeolite molecular sieve. structure, chemistry, and use. John Wiley & Sons, New York
Ashtekar S, Chilukuri SVV, Chakrabarty DK (1994) Small-pore molecular sieves SAPO-34 and SAPO-44 with chabazite structure. J Phys Chem, https://doi.org/10.1021/j100069a018
Aghamohammadi S, Haghighi M (2015) Dual-template synthesis of nanostructured CoAPSO-34 used in methanol to olefins. Chem Eng J, https://doi.org/10.1016/j.cej.2014.11.102
Wang P, Lv A, Hu J, Xu J, Lu G (2012) The synthesis of SAPO-34 with mixed template and its catalytic performance for methanol to olefins reaction. Microporous Mesoporous Mat, https://doi.org/10.1016/j.micromeso.2011.11.037
Izadbakhsh A, Farhadi F, Khorasheh F, Sahebdelfar S, Asadi M, Yan ZF (2009) Key parameters in hydrothermal synthesis and characterization of low silicon content SAPO-34 molecular sieve. Microporous Mesoporous Mat, https://doi.org/10.1016/j.micromeso.2008.12.009
Blackwell CS, Patton RL (1988) Solid-state NMR of silicoaluminophosphate molecular sieves and aluminophosphate materials. J Phys Chem, https://doi.org/10.1021/j100324a055
Izadbakhsh A, Farhadi F, Khorasheh F, Sahebdelfar S, Asadi M, Feng YZ (2009) Effect of SAPO-34’s composition on its physico-chemical properties and deactivation in MTO process. Appl Catal A Gen, https://doi.org/10.1016/j.apcata.2009.05.022
Dumitriu E, Azzouz A, Hulea V, Lutic D, Kessler H (1997) Synthesis, characterization and catalytic activity of SAPO-34 obtained with piperidine as templating agent. Microporous Mater, https://doi.org/10.1016/S0927-6513(96)00107-1
Acknowledgements
The authors acknowledge the financial support for this research by the Foundation for Research Support of the State of Amazonas (FAPEAM) and the Coordination for the Improvement of Higher Education Personnel (CAPES).
Funding
Foundation for Research Support of the State of Amazonas (FAPEAM) and Coordination for the Improvement of Higher Education Personnel (CAPES).
Author information
Authors and Affiliations
Contributions
WLA and GPV provided the idea for the research. All authors wrote, read, and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
dos Anjos, W.L., Morales, S.A.V., Oliveira, N.M.B. et al. Effect of silica/alumina ratio and structure-directing agent on the physical and chemical properties of SAPO-34. J Sol-Gel Sci Technol 100, 466–476 (2021). https://doi.org/10.1007/s10971-021-05669-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10971-021-05669-w