
Abstract
Vegetable oils are attractive substrates for use as renewable feedstock in polymer industry. Hydrosilylation is a way to insert silicon groups in an olefin, and if the silylated olefin has hydrolysable groups, it can be used as precursor reagent in sol–gel synthesis. In this work, for the first time, the hydrosilylation of the vegetable soybean oil is reported, and it was used as sol–gel molecular precursor, along with tetraethylorthosilicate, varying their proportions. The obtained organic–inorganic silica-based hybrid material was characterized by infrared spectroscopy, N2 adsorption–desorption isotherms, scanning electron microscopy, and thermogravimetric analysis. The hybrid contains the oil chain covalently anchored, and it presents interesting textural characteristics, such as high surface area and mesoporosity, which seem not be markedly affected by the organic amount added.
Graphical Abstract

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The search for sustainable chemical products has experienced an enormous grow in the last few years. In this sense, vegetable oils are very promising substrates for a wide scope of applications such as biodiesel, lubricants, biopolymers, coatings, and paints along with other applications [1–7]. Vegetable oils are easily available, non-toxic, biodegradable, and present low cost. They are constituted of saturated, mono-, and polyunsaturated fatty acid residues linked by glycerol, forming triglycerides. The distribution of even-numbered carbon chains depends on many factors, as for example the plant from which it has been obtained, the weather, and the soil conditions. There is prevalence of C16–C18 chains, either unsaturated or saturated, C18:1 (18 carbons length, one unsaturation, oleic acid) being the most often occurring chain [7, 8].
Oilchemistry has two major branches. The first consists in converting glycerol into valuable products [9], and the second deals with transformations on acid residues [10]. These can be performed in two different positions: in the carboxyl groups (yielding fatty esters, alcohols, amides and other derivatives) or in the organic chains [11]. This last one has been subject of studies in the last years. Triglycerides or their fatty esters have been reported as substrates for metathesis [12, 13], epoxidation [14–16], and hydroformylation [17–19] reactions.
Addition of silanes (e.g., dimethylchlorosilane HSiMe2Cl or triethoxysilane HSi(OEt)3) in the organic chain of the unsaturated fatty acids can be achieved by hydrosilylation reaction [10]. The catalytic addition of hydrogen by silicon hydrides to multiple bonds (reported as hydrosilylation or hydrosilation), in particular to C=C or C≡C bonds, has proved to be an efficient method for the formation of organosilicon compounds. The catalytic hydrosilylation has been used for crosslinking silicone polymers to elastomers and silicone-based coatings, and also for coupling of silanes and siloxanes to organic polymers. In this way, it is a reaction widely used in the silicone industry [20].
First results concerning the addition of silicon hydrides to internal unsaturated fatty acid esters were published in 1955 by Speier [21] followed by a few others [22–27]. Homogeneous catalyst based on transition metal complexes, especially Co(I), Rh(I), Pd(0), and Pt(0) compounds are the preferred used catalysts for hydrosilylation. The reactivity of terminal unsaturated fatty acid esters is higher than that of internal unsaturated ones [22–24]; for example, the hydrosilylation of 10-undecenoate, which has a terminal double bond, gave a higher yield than that of methyl oleate. This can constitute a difficulty when vegetable oils are used, since all its double bonds are internal. By this reason, one can explain why there are very few studies dealing with natural oils or its fatty esters as substrates for hydrosilylation [27, 28].
In the last decade, there has been wide interest in organic–inorganic hybrid materials. These materials present characteristics of both organic and inorganic components and even new characteristics [29]. In this context, vegetable oil silica-based hybrid materials as films of hydroxylated or epoxidized soybean oil have been reported [30], as well as hybrid materials using hydrosilylated fatty compounds [28, 31, 32]. However, as far as we know, there are no studies concerning the hydrosilylation of soybean oil and its sol–gel polymerization.
In this work, for the first time, the hydrosilylation of soybean oil is described. The hydrosilylated soybean oil was characterized and used as sol–gel precursor, along with tetraethylorthosilicate, for the synthesis of a novel mesoporous organic–inorganic hybrid material. The hybrid was characterized by thermogravimetric analysis, N2 adsorption–desorption isotherms, infrared spectroscopy, and scanning electron microscopy.
2 Experimental section
2.1 Materials
Soybean oil was supplied by Oleoplan, Ltda. (Veranópolis, Brazil). It was used after passing through silica gel column and degassed under vacuum. All other reagents were available from chemical suppliers and used as received. Wilkinson’s catalyst (Rh(PPh3)3Cl), soybean oil, and triethoxysilane were stored under argon atmosphere.
2.2 Soybean oil hydrosilylation
In a typical reaction, Wilkinson’s catalyst (30 mg, 3.24 × 10−2 mmol) was used with appropriate amounts of soybean oil and triethoxysilane based on the desired molar ratio between these reactants. The reactions were carried out under magnetic stirring and argon atmosphere using Schlenk’s techniques [33] at three set temperatures (80, 100, and 120 °C) for 24 h. After that, the remaining unreacted silane was removed under vacuum. The oil was then passed through silica for catalyst removal, and traces of remaining silane were removed again under vacuum until no Si–H infrared peak (2200 cm−1) could be observed.
2.3 Hydrosilylated soybean oil characterization
Soybean oil and hydrosilylated soybean oil were submitted to 1H nuclear magnetic resonance (1H NMR), infrared (FTIR), and thermogravimetric analyses (TGA). The 1H NMR spectra were measured using a Varian VNMRS 300 MHz spectrometer. The FTIR spectra were measured in Bruker Alpha-P FTIR/ATR equipment, with 32 scans. The TGA were performed using a TA Instruments SDT Q600 under nitrogen atmosphere. An average sample weight of 5 mg was heated in the temperature range of 25–700 °C, with a heating rate of 10 °C min−1. From 1H NMR [34], the soybean oil presents an average molar weight of 932.48 g mol−1 and an average number of double bonds per triglyceride unity of 4.7. Its density was determined as 0.8937 g mL−1.
2.4 Calculation of the hydrosilylation degree
The TGA technique was used to calculate the hydrosilylation degree by quantifying the remaining silica, since a partial hydrosilylation in C=O groups changes the peaks that are used for its calculation using Miyake’s method [34]. Hydrosilylation is an addition reaction, so if s moles of silane are added to the oil, the average molecular weight of the new product is now (Eq. 1)
where \( M_{\text{SO}} \) is the average molecular weight of the starting oil and \( M_{\text{sil}} \) the molecular weight of the silane used.
When one mole of hydrosilylated oil is totally burned, s moles of silica are generated. TGA gives p, which is the percentual ratio of the mass of remaining silica and the mass of original sample. So, for 1 mol of hydrosilylated oil (Eq. 2)
where \( M_{{{\text{SiO}}_{2} }} \) stands for the molecular weight of silica. Isolating s, we find (Eq. 3)
As the degree of hydrosilylation H is given by the percentual ratio between the number of moles of remaining silica after TGA and the number of possible active sites for hydrosilylation x, it can be expressed as (Eq. 4)
For the particular soybean oil used in this work, x = 7.7 (4.7 C=C, determined by 1H NMR [34], plus 3 C=O double bonds per mole of triglyceride) and the average molecular weight was 932.48 g mol−1 (also determined by Myake’s method [34]). Since the molecular weight of triethoxysilane is 164.27 g mol−1, the previous equation becomes (Eq. 5)
This equation allows the determination by TGA of the hydrosilylation degree for each run.
2.5 Synthesis of hybrid materials
TEOS (10 mL, 9.33 g) was pre-hydrolyzed in the presence of methyl alcohol (5 mL), water (1.6 mL), isoamyl alcohol (2.5 mL), and two drops of HF (40 %). The mixture was kept under stirring for 10 min, at room temperature. Afterward, solutions containing 0.28 g (named S3, 3 % w/w organic content), 0.55 g (S6, 6 %), or 1.0 g (S10, 10 %) of hydrosilylated soybean oil, dissolved in 40 mL of isoamyl alcohol, were added along with five additional drops of HF. The mixture was gelified at 40 °C for 30 days. The resulting solid was comminuted, washed several times with water and ethanol, and dried in vacuum at 110 °C for 2 h. The reaction that describes the preparation of the xerogel is represented by Scheme 1.

Representation of hybrid material synthesis
2.6 Characterization of hybrid materials
Hybrid material samples were characterized by infrared (FTIR), N2 adsorption–desorption isotherms, scanning electron microscopy (SEM), and thermogravimetric analysis (TGA). The Fourier transform infrared spectroscopy (FTIR) analysis was performed using self-supporting disks with a diameter of 2.5 cm, weighing ca. 100 mg. The disks were heated for 60 min, at 140 °C, under vacuum. The IR cell used in this work was described elsewhere [35]. The spectra were obtained at room temperature in a Shimadzu FTIR Prestige 21 with 100 cumulative scans. The carbon elemental analyses were carried out in a CHN Perkin Elmer M CHNS/O Analyzer, model 2400. The SEM images were obtained by using a JEOL JSM 6060 microscope, operating at an acceleration voltage of 7 kV. Prior the analysis, samples were placed on a double-sided carbon tape on a sample holder, and they were coated with gold, under vacuum. The textural analysis was made using N2 adsorption–desorption isotherms, determined at liquid nitrogen boiling point, using Tristar 3020 Kr Micromeritics equipment. The samples were previously degassed at 120 °C under vacuum, for 12 h. The specific surface areas were determined by the Brunauer–Emmett–Teller (BET) multipoint technique, and the pore size distributions were obtained by using the Barret–Joyner–Halenda (BJH) method [36]. TGA were performed in a similar fashion as for the hydrosilylated soybean oil.
3 Results and discussion
3.1 Hydrosilylation of soybean oil
Soybean oil hydrosilylation was performed starting from crude soybean oil, triethoxysilane, and Wilkinson’s catalyst. The expected C=C hydrosilylation reaction is represented in Scheme 2.

Representation of soybean oil hydrosilylation reaction
The hydrosilylation reaction was evidenced by infrared analysis shown in Fig. 1. It is possible to observe, in the spectrum of crude soybean oil, the olefins bands at 3007 and 1654 cm−1, which are assigned to = C–H and –HC=CH– stretching bands (Fig. 1a) [37]. These bands seem to be less intense in hydrosilylated oil spectrum (Fig. 1b), indicating that double bonds were hydrosilylated. The apparent decrease in the intensity of stretching carbonyl band at 1743 cm−1, for hydrosilylated oil (Fig. 1b), indicates that the carbonyl group undergoes a partial hydrosilylation reaction too. It is also possible to observe in the hydrosilylated oil spectrum new bands with maximum near to 1077 and 964, assigned to Si–OR stretching and a band at 790 cm−1, which was attributed to Si–C stretching [37]. The bands at 2925 and 2855 cm−1 are due to C − H stretching of aliphatic chain. It is important to point out that the spectra do not show bands due to Si–H stretching, which appears in the range of 2100–2200 cm−1, showing that the unreacted silane was completely removed under vacuum. Bands which appear between 3700 and 3200 cm−1 and are due to silanol groups, SiO–H stretching [38], are not present as well, indicating that the reaction was carried out under total absence of moisture, preventing the hydrolysis of alcoxi groups.

Infrared spectra: a crude soybean oil and b hydrosilylated soybean oil. The bar value is 0.2
The hydrosilylation was also confirmed by 1H NMR analysis (Fig. 2), where it is possible to see in the spectrum of hydrosilylated oil a new group of peaks around 3.8 ppm, which was assigned to the presence of Si(OEt)3 moiety and also a decrease in the C=C peaks (5.4 ppm), proving the hydrosilylation reaction. The glycerol hydrogens near to 4.2 ppm had also been changed, and this feature can be interpreted as a consequence of carbonyl group hydrosilylation. Additionally, the peaks around 2.8, 2.3, 2.0, and 1.6 ppm, which are related to hydrogens near to double C=C bonds, show a decreasing in their intensity.

NMR spectra: a crude soyben oil and b hydrosilylated soybean oil
As discussed above, the hydrosilylation took place not only in C=C bonds but also in a fraction of carbonyl groups. In this way, the glycerol hydrogens, used for estimating the reaction yields by 1H NMR on carbon chains [34, 39, 40], cannot be applied here. Because of this, TGA were used to determine the hydrosilylation degree, in order to choose the best catalytic condition, as described above, since TGA of crude soybean oil gave no residue after it has been heated up to 600 °C. Note that if a double bond is converted in a non-silicon species (e.g., by hydrogenation), this transformation is not considered by this method. The hydrosilylation degree was assigned as H, and its values for different reaction conditions are summarized on Table 1.
When 0.25 % catalyst was used (Entry 1, Table 1), a partial hydrosilylation was achieved (H = 27 %). For 0.5 % catalyst (Entry 2, Table 1), a better result was obtained (H = 44 %). Attempts to improve H values by increasing or decreasing the temperature showed the opposite effect (Entry 3 and 4, Table 1) (H ~ 20 %). As well, lower amounts of silane decrease the hydrosilylation degree (Entry 5, Table 1). When a larger scale was applied, even for 0.5 % catalyst, H decreased to 22 % (Entry 6, Table 1). Thus, knowing that the chains of oil have 7.7 double bonds on average (4.7 C=C, as discussed in Sect. 2.3, and 3 C=O), it is possible to state that statistically most of the chains are hydrosilylated in at least one site. Therefore, it can be inferred that the soybean oil was successfully hydrosilylated, and the product of the large-scale reaction (Entry 6, Table 1) was used as organic precursor for sol–gel synthesis with TEOS, to obtain the hybrid material containing anchored soybean oil.
3.2 Hybrid material
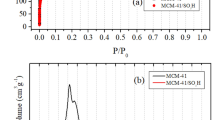
The hybrid material samples were characterized by N2 adsorption–desorption analysis. The isotherms and pore size distributions are shown in Fig. 3, and the textural characteristics are presented in Table 2, along with carbon elemental analysis. It was observed a small decrease in the surface area with the organic amount increasing, and this behavior was already reported [41, 42]. As it can be seen in Fig. 3a, all samples have isotherms of mesoporous materials (type IV isotherms) [36]. This feature can be interpreted by the use of fluoride as catalyst in the hydrolysis and condensation sol–gel reactions. It was reported that nucleophilic fluoride catalyst leads to the formation of spherical primary particles. The xerogel produced by the interconnection of these primary particles presents mesoporous structure [43, 44]. In Fig. 3b, it is possible to see that the samples S3 and S6 showed a very similar pore size distribution with a maximum around 9 nm, while the S10 sample presents a broader distribution, and it is shifted for higher values, with maximum around 11 nm. It was also reported that pore size of silica-based hybrid materials is influenced by the organic precursor amount added in the synthesis. In a general way, if the organic amount is greater, the smaller pores are obtained [41, 42]. However, in this work, an opposite effect was observed for S10 sample.

Textural analysis: a N2 adsorption–desorption isotherms b BJH pore size distribution
The TGA of hybrid material samples are shown in Fig. 4, and two main weight losses can be observed: first from room temperature up to 120 °C, due to water desorption and a second one from 120 °C until 600 °C, which was assigned to the organic desorption by thermal decomposition. The amounts of water and organics are summarized on Table 3. It is possible to see on Table 3 that the water amount decreases from S3 to S10 samples. This result was interpreted by taking into account the hydrophobicity of the organic moiety. The greater the organic quantity added in the synthesis, the lower the amount of water adsorbed on surface of material. Regarding the weight loss above 120 °C, due to the organic desorption, the results agree with the carbon elemental analysis shown on Table 2 and also with the planned synthesis. Although the organic quantity obtained from TGA (Table 3) was higher than those found from elemental analysis (Table 2), even when taking the hydrogens masses into account, it is important to point out that the TGA weight loss values have also contribution of silica dehydroxilation [38, 41], which occur along with the whole thermal treatment.

TGA of hybrid material samples
The hybrid material samples were also characterized by infrared spectroscopy (Fig. 5). It is possible to see typical spectra of hybrid material, with bands due to organic and also inorganic components. The inorganic silica can be identified by the typical overtone bands with maximum around 1870 cm−1 and the free silanol stretching at 3740 cm−1 [38]. The organic moiety is clearly identified by bands at 2930 and 2855 cm−1, due to C − H stretching of aliphatic chain, also bands at 1704 and 1465 cm−1, assigned to C − O stretching and CH2 bending, respectively [37]. These bands remain in the spectra, even after thermal treatment at 140 °C, indicating that the organic moiety is covalently bonded [35], corroborating that the hydrosilylation was successfully attained. These organic bands seem to increase in their intensities from S3 to S10 samples, when compared with the silica overtone band, which remains constant for all samples. Another interesting feature of these spectra is the band around 1630 cm−1, which is due to the bending of water [38]. This band seems more evident for samples with higher organic content, S6 and S10 samples. It is important to point out that the samples were previously submitted to thermal treatment in vacuum, at 140 °C, and the spectra were obtained without exposing the sample to the external atmosphere. In this way, the presence of this band could indicate the presence of remaining traces of water trapped inside the pores, probably due to the hydrophobicity of the organic long chains.

Room-temperature infrared spectra of hybrid material samples, previously heated at 140 °C, for 1 h, under vacuum. The bar value is 0.5
The hybrid material samples were investigated by scanning electron microscopy (SEM), and the images are shown in Fig. 6. It can be observed that the materials have a bead-shaped morphology, with some aggregates, although no significant changes could be observed between the hybrid materials, considering the oil–TEOS ratios studied and the observed magnification.

Scanning electron microscopy (SEM) images of the hybrid materials with different oil–TEOS ratio: a Sample S10, b sample S6, and c sample S3
4 Conclusions
Soybean oil was successfully hydrosilylated with triethoxysilane using Wilkinson’s catalyst. The obtained hydrosilylated soybean oil was used as precursor in the synthesis of hybrid material, using sol–gel method, varying the TEOS and hydrosilylated soybean oil proportions. The organic component is strongly bonded to the silica matrix, presenting good thermal stability. All hybrid samples showed mesoporous structure, with pore diameter around 10 nm. The increase in the organic amount did not produce significant changes in the textural characteristics.
References
Biermann U, Bornscheuer U, Meier MAR, Metzger JO, Schäfer HJ (2011) Angew Chem Int Ed Engl 50:3854–3871
Winkler M, Lacerda TM, Mack F, Meier MAR (2015) Macromolecules 48:1398–1403
Balbino JM, de Menezes EW, Benvenutti EV, Cataluña R, Ebeling G, Dupont J (2011) Green Chem 13:3111–3116
Alam M, Akra D, Sharmin E, Zafar F, Ahmad S (2014) Arabian J Chem 7:469–479
Miao S, Wang P, Su Z, Zhang S (2014) Acta Biomater 10:1692–1704
Sharma RV, Somidi AKR, Dalai AK (2015) J Agric Food Chem 63:3235–3242
Deuss PJ, Barta K, de Vries JG (2014) Catal Sci Technol 4:1174–1196
Behr A, Westfechtel A, Gomes JP (2008) Chem Eng Technol 31:700–714
Behr A, Eilting J, Irawadi K, Leschinski J, Lindner F (2008) Green Chem 10:13–30
Huber T, Firlbeck D, Riepl HM (2013) J Organomet Chem 744:144–148
El Kadib A, Asgatay S, Delpech F, Castel A, Rivière P (2005) Eur J Org Chem 21:4699–4704
Öztürk BO, Topoglu B, Şehitoglu SK (2015) Eur J Lipid Sci Technol 117:200–208
Baibich IM, Gregório JR, Kern C, Rudler H (1996) J Braz Chem Soc 7:83–86
Riley SJ, Verkade JG, Angelici RJ (2015) J Am Oil Chem Soc 92:589–601
Gerbase AE, Brasil MC, Gregório JR, Mendes ANF, von Holleben MLA, Martinelli M (2002) Grasas Aceites 53:175–178
Gregório JR, Gerbase AE, Martinelli M, Brasil MC, Mendes ANF (2002) J Am Oil Chem Soc 79:179–181
Behr A, Vorholt AJ (2012) In: Meier MAR, Weckhuysen BM, Bruijnincx PCA (eds) Hydroformylation and related reactions of renewable resources, topics in organometallic chemistry, vol 39. Springer, Berlin, pp 103–128
Mendes ANF, da Rosa RG, Gregório JR (2005) Catal Commun 6:379–384
Mendes ANF, Gregório JR, da Rosa RG (2005) J Braz Chem Soc 16:1124–1129
Troegel D, Stohrer J (2011) Coord Chem Rev 255:1440–1459
Speier JL, Zimmerman R, Webster J (1956) J Am Chem Soc 78:2278–2281
Saghian N, Gertner D (1974) J Am Oil Chem Soc 51:363–367
Behr A, Naendrup F, Obst D (2002) Adv Synth Catal 344:1142–1145
Behr A, Naendrup F, Obst D (2002) Eur J Lipid Sci Technol 104:161–166
El Kadib A, Katir N, Castel A, Delpech F, Rivière P (2007) Appl Organomet Chem 21:590–594
El Kadib A, Castel A, Delpech F, Rivière P (2005) Chem Phys Lipids 148:112–120
Delpech F, Asgatay S, Castel A, Rivière P, Rivière-Baudet M, Amin-Alami A, Manriquez J (2001) Appl Organomet Chem 15:626–634
Lligadas G, Ronda JC, Galià M, Cádiz V (2006) Biomacromolecules 7:2420–2426
Benvenutti EV, Moro CC, Costa TMH, Gallas MR (2009) Quim Nova 32:1926–1933
Brasil MC, Gerbase AE, de Luca MA, Gregório JR (2007) J Am Oil Chem Soc 84:289–295
Lligadas G, Callau L, Ronda JC, Galià M, Cádiz V (2005) J Polym Sci A Polym Chem 43:6295–6307
El Kadib A, Katir N, Marcotte N, Molvinger K, Castel A, Rivière P, Brunel D (2009) J Mater Chem 19:6004–6014
Shriver DF, Drezdzon MA (1986) The manipulation of air-sensitive compounds, 2nd edn. Wiley, New Jersey
Miyake Y, Yokomizo K, Matsuzaki N (1998) J Am Oil Chem Soc 75:15–19
Pavan FA, Gobbi SA, Costa TMH, Benvenutti EV (2002) J Therm Anal Calorim 68:199–206
Gregg SJ, Sing KSW (1982) Adsorption, surface area and porosity, chap 3 and 4. Academic Press, London
Colthup NB, Daily LH, Wiberley SE (1975) Introduction to infrared and Raman spectroscopy, 2nd edn, chap 7, 10 and 12. Academic Press, New York
Costa TMH, Gallas MR, Benvenutti EV, da Jornada JAH (1997) J Non-Cryst Solids 220:195–201
Gregório JR, Mendes ANF, da Rosa RG (2012) Quim Nova 35:1940–1944
Gregório JR, da Rosa RG, Mendes ANF, Bayón JC (2011) Catal Lett 141:977–981
de Menezes EW, Lima EC, Royer B, de Souza FE, dos Santos BD, Gregório JR, Costa TMH, Gushikem Y, Benvenutti EV (2012) J Colloid Interf Sci 378:10–20
Arenas LT, Vaghetti JCP, Lima EC, Moro CC, Benvenutti EV, Costa TMH (2004) Mater Lett 58:895–898
Benvenutti EV, Moro CC, Costa TMH, Gallas MR (2009) Quim Nova 32:1926–1933
Pavan FA, Gobbi SA, Moro CC, Costa TMH, Benvenutti EV (2002) J Porous Mater 9:307–311
Acknowledgments
The authors thank to CNPq, CAPES, FAPERGS, and PROPESQ/UFRGS for financial support and grants. This work was also supported by PRONEX/CNPq/FAPERGS-04/0887-0 and 10/0050-6. The authors thank also the CME-UFRGS for SEM images.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Fuscaldo, R.d.S., de Menezes, E.W., Lima, M.F.S. et al. Mesoporous organic–inorganic hybrid material containing hydrosilylated soybean oil. J Sol-Gel Sci Technol 78, 457–464 (2016). https://doi.org/10.1007/s10971-016-3957-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10971-016-3957-8