
Abstract
Comparisons of statistical analysis results between five methods using gamma spectrometry and a control group for measuring the radioactivity of uranium were performed to suggest the optimal method. In the statistical tests including linear regression, and Pearson’s correlation all gamma spectrometry methods correlated with the control group method though they had each disadvantage. The Student’s t-test results for a novel method 5 including a 230Th contribution, were 0.986 (235U) and 1.821 (238U), respectively. It had the advantage of being more accurate when evaluating the activity of 235U and 238U simultaneously. The novel method 5 can thus be recommended over others.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Accurate measurement of uranium radioactivity series in samples is important for estimating individual or public doses based on environmental radiation [1, 2]. According to a report by the United Nations Scientific Committee on the Effects of Atomic Radiation, approximately 50% of the worldwide average annual effective dose (2.4 mSv y−1) is from radon inhalation, and 21% is from terrestrial gamma rays such as 238U, 232Th decay series, and 40K [3]. These radionuclides mainly exist in soil, and they can be used for the characterization of samples and to distinguish the origin of samples [4]. In dose assessment studies, accurately measuring the uranium and thorium activities in the soil is essential because it is known that the uranium and thorium contribution to the dose is relatively high [5, 6]. The radioactivity of these nuclides in the soil can be measured with several methods, such as mass spectrometry [7,8,9,10,11], alpha-ray spectrometry, and gamma-ray spectrometry [9, 12].
One of the methods for determining uranium activity, considered in this study, is gamma spectrometry, which primarily measures and analyzes the peak counts originating from gamma-ray emissions in the samples. For instance, the high purity germanium (HPGe) detector is widely used to measure the uranium activity of a sample in monitoring environmental radiation because of its good energy resolutions and minimized pretretments [13, 14].
The gamma-ray measurement methods to analyze the radioactivity of uranium in the soil can be broadly classified into three types, assuming secular equilibrium of radionuclides. The first is to use a single energy peak emitted from a radionuclide (uranium progeny) in a sample. For instance, radionuclides and emission gamma-ray energy used to determine the 238U activity are 234Th (63.3 keV), 214Pb (351.9 keV), 214Bi (609 keV), and 234mPa (1001 keV) [14,15,16,17]. The second type for evaluating the activity of 235U and 238U is to use correction factors for a186 keV peak, which are based on the natural abundances of uranium isotopes [18, 19]. In the third type, multiple gamma-rays emitted from the 238U daughters, such as 144 and 186 keV, are used to determine the radionuclide activity. For instance, the identified peaks in the spectra can be a combination of peaks emitted by several radionuclides, e.g., the 186 keV peak is the combination peak of 226Ra and 235U in the soil [19,20,21,22]. These methods, however, have some limitations and problems such as emission intensities of the gamma-ray, the low detection efficiency of a detector, analysis uncertainty, and a coincident summing effect. For example, the emission intensity of the 1001 keV gamma-ray of 234 mPa is as low as 0.8435%, thus determining the uranium activity using the 1001 keV should be very carefully performed [23].
The method for determining uranium activity for a control group is the alpha spectrometry that assess 235U, 238U, and radionuclides of the uranium decay series [7, 24, 25]. Although this method requires complex chemical processes including incineration, acid dissolution, and electrochemical deposition [26, 27], the alpha spectrometry is known to have high accuracy with low background noise.
In this study, A novel method, which reflects the 230Th contributions in 144 and 186 keV peaks, is proposed to determine the precise radioactivity of the 235U and 238U. Analysis results of the five methods, which included the novel method were compared with alpha spectrometer results for 26 soil samples. In addition, this study discusses the validity of the methods for evaluating the radioactivity of the 235U and 238U for regular survey samples of environmental radiation monitoring.
Experimental
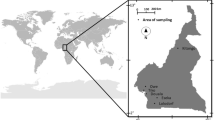
In this study, 26 soil samples were obtained from 0 to 5 cm below the ground surface in Gyeongju city, Republic of Korea, between April 2016 and April 2021, as shown in Fig. 1. After the soil samples were dried and pulverized, rocky and biological materials of large size were removed using a 2-mm mesh sieve.

Soil sampling locations in Gyeongju city, Republic of Korea
After incineration (450 °C and 48 h) for alpha spectrometry, a few portions of the dry samples were chemically pretreated including a tracer (232U). Dissolution using mixed acid (HF, HNO3, and HClO4, 1:4:1, respectively), and separation of iron and uranium using a UTEVA resin were performed [26,27,28]. The treated samples were deposited electrochemically on a plate for measurements with an alpha spectrometer (Alpha duo, ORTEC, USA) for 100,000 s [7]. The detection efficiency of the alpha spectrometer was corrected using an alpha-emitting 232U tracer (5263 and 5320 keV). The minimum detectable activity (MDA) of 235U and 238U with the alpha spectrometer was determined to be about 0.10 and 0.3 Bq kg−1, respectively. The results from alpha spectrometry were applied as a control group for a statistical comparison with gamma spectrometry results in this study.
Soil samples for measurements using a gamma spectrometer were placed in a 450 mL Marinelli beaker with a silicone cover. The soil samples were measured for nearly one day (live time: 86,000 s), and nine p-type HPGe detectors in a radiation research laboratory were randomly used during the radiation monitoring. The measurement results were analyzed using GENIE 2000 or an Aptec Multichannel Analyzer. A nuclide library file (eml300.lib) was used for nuclide identification. Detection efficiency of these HPGe detectors was determined using a certificated reference material of the Korea Research Institute of Standards and Scienc, including 214Am (59.54 keV), 109Cs (88.03 keV), 57Cd (122.06 keV, 135.47 keV), 139Ce (165.86 keV), 51Cr (320.08 keV), 113Sn (391.70 keV), 85Sr (514 keV), 137Cs (661.7 keV), 60Co (1173.23 keV, 1332.49 keV), and 88Y (898.04 keV, 1836.05 keV). The detection efficiency was corrected for self-absorption related to the density [29,30,31].
The measurements of the real soil samples were performed without waiting for radon secular equilibrium in the Marinelli beaker; however, the uranium activities in the soil samples were determined under the assumption of secular equilibrium, including radon gas. The uncertainty of the measurement results could thus be increased becuase of the nonequilibrium influence of chemical reactions and biological interactions of materials in the soil, which was taken into consideration [16]. The MDA of the gamma spectrometry was assessed referring to the Currie method and a recommended method of ISO-11929. [32, 33].
The first type of activity evaluation was a single peak analysis that used a single gamma-ray line emitted from a radionuclide, in some cases with corrections from other radionuclide contributions. The second activity evaluation type used correction factors based on an abundance of natural uranium (235U and 238U) [18, 19]. The third activity evaluation type was a multiple peak analysis method, which evaluates radioactivity considering two or more energy peaks simultaneously, such as the 144 and 186 keV peaks [20, 21]. Table 1 shows summaries of the radioactivity determination methods and the main energy peak used at assessment. Table 2 lists the radionuclides contributing to the peaks at 144 and 186 keV.
Single peak analysis method
The single peak analysis method used the most prominent energy peak of the radionuclide to determine the radioactivity. In this study, two methods using different energy (method 1 (63.6 keV) and method 2 (352 keV)) were applied only to determiate the 238U activity, because a prominent peak worth consideration cannot be found for the 235U radionuclide.
In method 1, 238U is determined by the activity of 234Th (63.3 keV, 3.765%) [34]. Its main emission peak subtracts the contribution of 232Th (63.8 keV, 0.263%) [34], which is evaluated from 228Ac (911 keV, 25.8%) [37]. As 234Th is the first progeny of 238U, method 1 could be used for evaluating the 238U activity, when assuming secular equilibrium [14, 40]. An average of the MDAs of method 1 using the Currie method was 16.17 ± 12.02 Bq kg−1.
In method 2, the 238U activity is determined via the activity of 214Pb (352 keV, 35.6%) [36], which is one of the progenies of 222Rn, assuming radioactive equilibrium in a perfectly sealed container, unlike the actual situation. With the probable leakage of radon gas, it was expected that the measurements with this method would be lower than the real value [16]. Nevertheless, we carried out the activity evaluation of method 2 for a comparison. The measured activities with methods 1 and 2 can be calculated using Eq. (1).
where A denotes an activity concentration (Bq kg−1) of a radionuclide; CNet is a net count of an analysis peak (the net count when all necessary subtractions have already been done); T is measurement time (s); m is a sample mass (kg); e is full energy peak detection efficiency, and Ig denotes an emission probability of the specific gamma-ray. The MDA average of method 2 using the background of the 352 keV ROI was 0.63 ± 0.17 Bq kg−1.
Correction factor method
The correction factor method (method 3) evaluates the activities of 235U and 238U. This method evaluates the 226Ra activity from the 186 keV peak net count and a correction factor (0.583 ± 0.01) based on the uranium natural abundance. Similarly, the 235U activity measurements are obtained by applying another correction factor (0.417 ± 0.01) [18, 19]. Equation (2), a slight modification to Eq. (1), is used to evaluate the activities of 235U or 238U.
where C186 is a net count of 186 keV peak, f nuc. is a correction factor of nuclides; i.e., 0.583 for 238U and 0.417 for 235U. However, it should be noted that the uranium abundance that is a basic assumption of method 3 may vary by region, as indicated in the study by Di Lella [41,42,43]. The MDA average of 235U and 238U of method 3 using the background of the 186 keV were 0.81 ± 0.24 Bq kg−1 and 3.36 ± 0.96 Bq kg−1, respectivtly.
Multiple peaks analysis method
The multiple peaks analysis method uses two or more gamma peaks emitted from several nuclides at the same time. In this study, to provide accurate radioactivity, the 144 keV and 186 keV peaks were corrected 228Ac (145.85 keV, 184.54 keV) contributions. Moreover, the 228Ac contribution to 144 keV and 186 keV peaks can be easily evaluated from a 911 keV energy peak.
Method 4 was previously published by other researchers; it uses a relation formula of radionuclides, which emits gamma rays near 186 keV and 144 keV. The formula was derived from the relation of 226Ra (186.21 keV), 223Ra (144.23 keV, 323.87 keV), and 235U (143.76 keV, 185.7 keV), assuming secular equilibrium [20, 21].
Method 5 is a suggestion method to evaluate the activity of 235U and 238U using a relation formula, similar to method 4, however, method 5 includes a correction for the activity of 230Th (143.87 keV, 186.05 keV). 230Th is the fifth progeny of 238U, and the emission gamma rays of this radioisotope could influence the radioactivity evaluation of 235U and 238U, considering the ratio of 235U and 238U. Becuase 235U (143.76 keV), 230Th (143.87 keV), and 223Ra (144.23 keV) gamma lines are very close, they usually form one single peak at 144 keV. The region of interest (ROI) of the 144 keV peak was roughly selected from 142 to 147 keV to reduce the analysis process uncertainty because it can be identified as a peak including 228Ac (145.85 keV), as shown in Fig. 2. We observed a broadened ROI for 11 of 26 cases. Similarly, the 228Ac (184.54 keV), 235U (185.7 keV), 230Th (186.05 keV), and 226Ra (186.21 keV) peaks were combined to form the 186 keV peak [37].

Gamma lines contributing to the144 keV peak: (1) 223Ra, (2) 230Th, (3) 235U, and (4).228Ac
The united count can be expressed as the count relation formula, including each radionuclide. Counting equations of 144 and 186 keV in broadened ROI cases are shown as follows:
where Ca is a net count of a broad ROI. A superscript and a subscript of \({C}_{a}^{b}\) are the mass number of a nuclide and gamma radiation energy (keV) emitted from the nuclide, respectively. For instance, 223, 226, 228, 230 and 235 denote 223Ra, 226Ra, 228Ac, 230Th, and 235U, respectively. The formulas corrected for the 228Ac contribution in the peak ROI are as follows:
where C*denotes a net count of the ROI after subtracting the 228Ac counts; Ab is the radioactivity (Bq kg−1) of’b’ nuclide in the soil sample. If the uranium series are in secular equilibrium (A235 equals A223, A226 equals A230), the radioactivity-count relation formulas become the followings:
where ε144 and ε186 denote the detection efficiency for the 144 and 186 keV energy peaks, respectively; and \({I}_{a}^{b}\) is the gamma-ray intensity of the radionuclides [34,35,36,37,38]. A system of linear Eq. (7) and Eq. (8) in which the radionuclide activity are variables is rearranged for the count. Equation (9) for the 235U activity is obtained from Eq. (7), and this equation can be put into Eq. (8) and rearranged to get Eq. (10) with one variable, A226;
Final formulas regarding the 235U activity and the 226Ra (238U) activity with radionuclide gamma-ray emission rate thus are summarized in Eq. (11) and Eq. (12), as below [34,35,36,37,38];
Before the 230Th correction, the denominator correction value (0.03403) of Eq. (12) was 0.0359, and the corrected terms of Eq. (11) were 0 (numerator) and 0.1423 (denominator). It has been assumed that the detection efficiency of a unified peak is the same because the energy of the emission gamma-rays is close, and the uncertainty of the calibrated efficiency had been approximately 4%. In this study, the detection efficiency of the central energy of ROI was used. For example, the efficiency of 144 and 186 keV were 0.049952 and 0.026549, respectively, in the roughly searched peaks. The MDA averages of 235U and 238U of methods 4 and 5 using the background of the 144 and 186 keV were 1.24 ± 0.31 Bq kg−1 and 1.31 ± 0.33 Bq kg−1, 20.25 ± 5.00 Bq kg−1 and 21.80 ± 5.44 Bq kg−1, respectively.
The correlation, linearity, and statistical similarity were compared to select a suitable gamma-ray analysis method based on the analysis cases. First, as the correlation, a Pearson’s correlation coefficient was calculated between those of each method and the alpha spectrometry results. If the Pearson’s correlation coefficient is close to 1, the comparison subjects are considered correlated. Second, as for the linearity, a simple linear regression with a weighting factor from the data’s uncertainty, which is the inverse of a square value of the radioactivity uncertainty, was performed by the Origin pro 2021 program as the linearity check or a linear model fitting. Finally, as for the statistical similarity, a paired t-test (Student’s t-test) was performed to confirm the statistical similarity between each gamma spectrometry method and the alpha spectrometry (control group) [44]. In the Student’s t-test, the null hypothesis (H0) is that the comparison subjects are the same, and the alternative hypothesis (H1) is they are not the same, generally [44]. If the t-value is higher than a critical value, it supports a statistical difference. In other words, if the t-value of an analysis variable is smaller than other, the variable could be considered to be relatively closer to the control group than to the others. In this study, the control group for statistical comparisons is the alpha spectrometer results, the experimental group results are those from the gamma-ray analysis methods.
Results and discussion
In this study, the uncertainty of these averages was shown as a 95% confidence interval with the standard uncertainty of each average, including the uncertainty of each measurement value, and the error term for each dataset was estimated with a general statistical method (e.g., uncertainty propagation).
In the comparison of simple arithmetic statistics, the average of 238U radioactivity evaluated with the alpha spectrometry results was 30.29 ± 2.79 Bq kg−1. The averages of 238U radioactivity for the five gamma spectrometry methods ranged from 25.56 ± 2.13 Bq kg−1 to 32.16 ± 2.63 Bq kg−1. The average of 235U radioactivity evaluated with the alpha spectrometer was 1.39 ± 0.14 Bq kg−1. The averages of 235U radioactivity for the three evaluation methods ranged from 1.46 ± 0.31 Bq kg−1 to 1.58 ± 0.16 Bq kg−1. The mean of the 228Ac activity for the peak count correction was evaluated as 33.08 ± 4.30 Bq kg−1. The contribution average of the 228Ac on the broadened ROI were calculated as 23.3% ± 4.2% in the 144 keV peak and 1.1% ± 0.1% in the 186 keV peak.
238U radioactivity evaluation results
The results of the evaluation of 238U radioactivity are shown in Fig. 3. The 238U activity average of method 1 was 31.81 ± 4.19 Bq kg−1, which was consistent with the average of the alpha spectrometry results as expected. The standard deviation (SD) of the 238U radioactivity was 10.28 Bq kg−1. The contribution of the 63.8 keV peak (232Th) into the 63.3 keV peak was determined to be 8 ± 3%. Therefore, considering the 232Th activity is better to evaluate the 234Th activity more accurately. The Pearson’s correlation coefficient of method 1 was 0.66; and the Student’s t-test result was 1.359. A slope, the y-intercept, and the determinant coefficient of the linear regression were 1.09 ± 0.23 and −2 ± 7 Bq kg−1, and 0.48, respectively.

238U radioactivity comparison between alpha and gamma spectrometry results: a methods 1 and 2, b method 3, and c methods (4) and (5)
In method 2, assuming radioactive equilibrium in a leak-tight container, the radioactivity average of 238U was evaluated as 25.56 ± 2.21 Bq kg−1, which is only 84.39% of the alpha spectrometer results. The SD of method 2 was 5.47 Bq kg−1. As expected, the reason for the lower activity results in this study could to attribute to a non-airtight container and a lack of waiting time to reach secular equilibrium. The Pearson’s coefficient was 0.70. However, the slope was 0.7 ± 0.1, the intercept was 4 ± 4 Bq kg−1, and R2 of the simple linear regression was 0.48. The paired t-test value was 5.84. In method 2, because of the time problem and the expected radon gas leakage from the sample container, the radioactivity of the evaluation was lower by 15.61%, and method 2 was thus considered invalid. However, if the container is ensured to be airtight and the time is adequate, the results should be better than in this study, as reported in another study [45].
The 238U activity average with method 3 was 32.16 ± 2.73 Bq kg−1. The SD of method 3 was calculated to be 6.75 Bq kg−1. Although the Pearson’s correlation coefficient of method 3 was evaluated to be 0.77, the R2 of the linear regression result of this method was 0.59, the slope of the fitting was 0.9 ± 0.2, and the intercept was 4 ± 4 Bq kg−1. The t-value of the paired t-test was 2.636.
The average values of 238U radioactivity as evaluated by method 4 and method 5 were 29.99 ± 2.89 Bq kg−1 and 31.27 ± 3.01 Bq kg−1, respectively. The SDs of these were 6.98 Bq kg−1 and 7.27 Bq kg−1. Although the 230Th contribution to the 186 keV peak count was included in method 5, the 238U evaluation results of method 5 were roughly 4% higher than those of method 4. This is because the 230Th activity contribution caused the 144 keV peak count or the 235U activity to decrease. The Pearson coefficients were 0.68 in both methods 4 and 5. The R2 values of the linear regression were 0.46 in method 4 and 0.47 in method 5, the slopes were 0.9 ± 0.2, and the intercepts were 2 ± 6 Bq kg−1. The multiple peaks analysis results were correlated to those from the alpha spectrometry and linearity was observed. The paired t-test results were 0.307 and 0.968. Based on the results of this analysis, both methods 4 and 5 provided acceptable t-test results when evaluating the radioactivity of 238U.
235U radioactivity evaluation results
The results of the evaluation of the 235U activity are shown in Fig. 4. The analysis results are presented in Table 3. The 235U radioactivity average of method 3 was 1.46 ± 0.30 Bq kg−1, and the SD was 0.31 Bq kg−1. The Pearson’s correlation coefficient was evaluated to be 0.69. The R2 value of the linear regression was 0.47, the slope of the fitting was 0.7 ± 0.2, and the intercept was 0.4 ± 0.2 Bq kg−1. The paired t-test result was 1.742.

235U radioactivity comparison between alpha and gamma spectrometry results: a method 3, and b methods (4) and (5)
The average 235U activity values of method 4 and method 5 were 1.58 ± 0.16 Bq kg−1 and 1.47 ± 0.18 Bq kg−1, respectively, and SDs were 0.41 Bq kg−1 and 0.40 Bq kg−1, respectively. The 235U activity average difference resulting from the 230Th radionuclide correction was roughly 7%. In the 144 keV energy peak count, the meager gamma-ray intensity of the Th was offset to be effective, because the high contribution of the 230Th activity and a low emission rate of the 235U. The Pearson’s correlation coefficients of method 4 and method 5 were 0.80 and 0.79, respectively. The R2 values of the linear regression results were 0.64 and 0.61; the slopes were 1.1 ± 0.2; the intercepts were 0.0 ± 0.2 Bq kg−1 and 0.1 ± 0.3 Bq kg−1, respectively. The paired t-test values were 4.262 and 1.821. Based on the results of this analysis, method 5 was better than method 4 with evaluating the radioactivity of 235U.
Comparisons
Table 3 shows a summary of statistical analysis results of this study. The Pearson’s correlation coefficients between gamma spectrometry methods and alpha spectrometry were greater than 0.66. Excluding methods 2 and 3 that lacked linearity of the slope, methods 4 and 5 resulted in the best value, 0.68. The determination coefficients of linear regression analyses were 0.46 to 0.59 in the 238U cases and 0.47 to 0.64 in the 235U cases. The slopes of methods 4 and 5 were close to unity. In the Student’s t-test between gamma spectrometry methods and the control group, the t-values were assessed from 0.307 to 5.840 on both 238U and 235U. In the 238U activity, the t-values for methods 2 and 3 were higher than the critical value of a 95% level of confidence (2.055), and in the 235U activity, the t-value of method 4 was higher. Based on these comparison results, it could be regarded that the gamma-ray spectrometry method results were all correlated with the alpha spectrometry results. Considering the statistical results and the linear regression, method 5, which was novel in this study, thus showed advantages of being more accurate when evaluating 235U and 238U simultaneously.
There are some problems with method 1, in that radioactivity assessment using 63.3 keV is difficult because around the low energy peak the detection efficiency of a p-type HPGe detector is generally low and correction factors like a density correction are needed [40]. In this study the activties from method 2 are obtained without waiting for radon secular equilibrium. It is expected that the measured values of 238U is reduced for the radon gas leakage and broken equilibrium. Nevertheless, if the soil container is airtight and the waiting time is long enough for equilibrium, the results can be better than those obtained in this study, as reported in previous research [46]. Method 3 has a drawback in that the abundance ratio of natural uranium may show regional variations especially for enrichment-related regions [11]. The weakness of methods 4 and 5 is that the MDAs were higher than with other methods but, the MDAs of methods 4 and 5 would improve by 18% (235U) and 16% (238U) when an ROI range at a 144 keV peak was reduced by excluding 228Ac.
Conclusions
In this study, to confirm validity of the novel and traditional uranium activity evaluation methods, the authors presented analysis results and statistical comparison results of five gamma spectrometry methods for measuring the activity of 238U and 235U in environmental soil samples. While each method had weaknesses, e.g., an uncertainty problem, broken secular equilibrium by gas leakage, the natural abundance differences of uranium, and the high MDA, all gamma spectrometry results in this study were correlated with the control group method. In addition, based on the statistical evidence, method 5, which included a 230Th contribution into consideration, possessed the advantage of being more accurate when evaluating the activity of 235U and 238U simultaneously, as shown in Table 3. Method 5 can thus be recommended as a method for evaluating the activity 235U and 238U. It is expected that the results of this study would be useful in selecting a uranium analysis method with gamma spectrometry for general environmental radioactivity monitoring.
References
Saito K, Petoussi-Henss N, Zankl M (1998) Calculation of the effective dose and its variation from environmental gamma ray sources. Health Phys 74:698–706
Mehra R, Singh M (2011) Measurement of radioactivity of 238U, 226Ra, 232Th and 40K in soil of different geological origins in Northern India. J Environ Prot (Irvine, Calif) 02:960–966
United Nations Scientific Committee on the Effects of Atomic Radiation (UNSCEAR) (2000) Sources and effects of ionizing radiation/United Nations Scientific Committee on the Effects of Atomic Radiation. UNSCEAR 2000 Rep. I:1–17
Ntsohi L, Usman I, Mavunda R, Kureba O (2021) Characterization of uranium in soil samples from a prospective uranium mining in Serule, Botswana for nuclear forensic application. J Radiat Res Appl Sci 14:23–33
Saito K, Jacob P (1995) Gamma ray fields in the ari due to sources in the ground. Radiat Prot Dosimetry 58:29–45
Hertel NE, Eckerman KF, Bellamy MB, et al (2015) Federal Guidance Report 15: external exposure to radionuclides in soil, air, and water
El-Galy MM, Issa FA, Desouky OA et al (2011) Use of alpha spectrometry for analysis of U-isotopes in some granite samples. J Radioanal Nucl Chem 288:805–811
Cowan GA, Adler HH (1976) The variability of the natural abundance of 235U. Geochim Cosmochim Acta 40:1487–1490
Jeambrun M, Pourcelot L, Mercat C et al (2012) Potential sources affecting the activity concentrations of 238U, 235U, 232Th and some decay products in lettuce and wheat samples. J Environ Monit 14:2902–2912
Qiao J, Lagerkvist P, Rodushkin I et al (2018) On the application of ICP-MS techniques for measuring uranium and plutonium: a Nordic inter-laboratory comparison exercise. J Radioanal Nucl Chem 315:565–580
Weyer S, Anbar AD, Gerdes A et al (2008) Natural fractionation of 238U/235U. Geochim Cosmochim Acta 72:345–359
Mola M, Palomo M, Peñalver A et al (2013) Comparative study of different analytical methods for the determination of 238U, 234U, 235U, 230Th and 232Th in NORM samples (Southern Catalonia). J Environ Radioact 115:207–213
García-Talavera M (2003) Evaluation of the suitability of various γ lines for the γ spectrometric determination of 238U in environmental samples. Appl Radiat Isot 59:165–173
Papachristodoulou CA, Assimakopoulos PA, Patronis NE, Ioannides KG (2003) Use of HPGe γ-ray spectrometry to assess the isotopic compositiion of uranium in soils. J Environ Radioact 64:195–203
Hasan M, Bódizs D, Czifrus S (2002) A simplified technique to determine the self-absorption correction for sediment samples. Appl Radiat Isot 57:915–918
Lenka P, Jha SK, Gothankar S et al (2009) Suitable gamma energy for gamma-spectrometric determination of 238U in surface soil samples of a high rainfall area in India. J Environ Radioact 100:509–514
Sutherland RA, De Jong E (1990) Statistical analysis of 7-emitting radionuclide concentrations for three fields in southern saskatchewan, canada. Health Phys 58:417–428
Salman A, Ahmed Z, Allam K, El-Sharkawy S (2019) A comparative study for 235 U radioactivity concentration calculation methods in phosphate samples. Radiat Prot Environ 42:5. https://doi.org/10.4103/rpe.rpe_77_18
Ebaid YY, El-Mongy SA, Allam KA (2005) 235U-γ emission contribution to the 186 keV energy transition of 226Ra in environmental samples activity calculations. Int Congr Ser 1276:409–411
Dowdall M, Selnaes G, Gwynn JP, Davids C (2004) Simultaneous determination of 226Ra and 238U in soil and environmental materials by gamma-spectrometry in the absence of radium progeny equilibrium. J Radioanal Nucl Chem 261:513–521
Ceccatelli A, Katona R, Kis-Benedek G, Pitois A (2014) Measurement of 226Ra in soil from oil field: advantages of γ-ray spectrometry and application to the IAEA-448 CRM. Appl Radiat Isot 87:461–467
De Corte F, Umans H, Vandenberghe D et al (2005) Direct gamma-spectrometric measurement of the 226Ra 186.2keV line for detecting 238U/226Ra disequilibrium in determining the environmental dose rate for the luminescence dating of sediments. Appl Radiat Isot 63:589–598
Yücel H, Çetiner M, Demirel H (1998) Use of the 1001keV peak of 234mPa daughter of 238U in measurement of uranium concentration by HPGe gamma-ray spectrometry. Nucl Instrum Methods Phys Res Sect A Accel Spectrometers Detect Assoc Equip 413:74–82
Danesi PR, Bleise A, Burkart W et al (2003) Isotopic composition and origin of uranium and plutonium in selected soil samples collected in Kosovo. J Environ Radioact 64:121–131. https://doi.org/10.1016/S0265-931X(02)00043-7
Boulyga SF, Testa C, Desideri D, Becker JS (2001) Optimisation and application of ICP-MS and alpha-spectrometry for determination of isotopic ratios of depleted uranium and plutonium in samples collected in Kosovo. J Anal At Spectrom 16:1283–1289
Burns PA, Clarck DE, Claxton D, et al (1999) Technologies for remediation of radioactively contaminated sites. IAEA Tecdoc-1086 101
Ko YG, Lim J-M, Chung KH, et al (2015) Alpha spectrometry for determination of 238U, 235U, 234U, 232Th and 230Th in Soil Sample. In: transactions of the Korean nuclear society spring meeting. Korean Nuclear Society
Cheolu Kim, et. al. (2017) Environmental Radioactivity Survey in Korea, KINS/ER-28, Vol. 49
Gilmore GR (2008) Practical gamma-ray spectrometry, 2nd edn. New York, Wiley
Al-Tuweity J, Kamleh H, Said Al-Masri M, et al (2021) Determination of correction factor of self-absorption for lead-210 in environment samples using spike method. E3S Web Conf 234:1–4
Jang E-S, Gim Y-S, Lee S-Y (2017) Marinelli beaker measurement and self absorption correction and application for various environmental samples in Monte Carlo simulation. J Radiol Sci Technol 40:605–611
Currie LA (1984) Lower limit of detection : definition andi elaboration of a proposed position for radiological EHluent and environmental measurements. NUREG/CR-4007
The International Organization for Standardization (ISO) (2010) Determination of the characteristic limits (decision threshold, detection limit and limits of the confidence interval) for measurements of ionizing radiation - Fundamentals and application. ISO-11929:2010(E) 60
M.-M.Be, V.Chiste, C.Dulieu, et al (2010) Table of radionuclides (Vol. 5 - A = 22 to 244). Monogr. BIPM-5, vol.5. 5:7 to 12
M.-M.Be, V.Chiste, C.Dulieu, et al (2011) Table of radionuclides (Vol. 6 – A = 22 to 242). Monogr. BIPM-5, vol.6. 6:306
M.-M.Be, V.Chiste, C.Dulieu, et al (2008) Table of radionuclides (Vol. 4 – A = 133 to 252). Monogr. BIPM-5, Vol.4. 4:309
Abusaleem K (2014) Nuclear data sheets for A = 228. Nucl Data Sheets 116:163–262
Akovali YA (1993) Nuclear data sheets for A = 230. Nucl Data Sheets 69:155–208
LaMont SP, Gehrke RJ, Glover SE, Filby RH (2001) Precise determination of the intensity of 226Ra alpha decay to the 186 keV excited state. J Radioanal Nucl Chem 248:247–253
Huy NQ, Luyen TV (2004) A method to determine 238U activity in environmental soil samples by using 63.3-keV-photopeak-gamma HPGe spectrometer. Appl Radiat Isot 61:1419–1424
Di Lella LA, Nannoni F, Protano G, Riccobono F (2005) Uranium contents and 235U/ 238U atom ratios in soil and earthworms in western Kosovo after the 1999 war. Sci Total Environ 337:109–118
Stirling CH, Andersen MB, Potter EK, Halliday AN (2007) Low-temperature isotopic fractionation of uranium. Earth Planet Sci Lett 264:208–225
Hiess J, Condon DJ, McLean N, Noble SR (2012) 238U/235U Systematics in Terrestrial Uranium-Bearing Minerals. Science (80- ) 335:1610 LP – 1614
Student (1908) The probable error of a mean. Biometrika 6:1.
Paiva JDS, Farias EEG, Franca EJ De (2015) Assessment of the equilibrium of Th-228 and Ra-228 by gamma-ray spectrometry in mangrove soils. In: INAC 2015: international nuclear atlantic conference Brazilian nuclear program State policy for a sustainable world
Lim S, Syam NS, Maeng S, Lee SH (2021) Determination of 226Ra in TENORM sample considering Radon leakage correction. J Radiat Prot Res 46:127–133
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Maeng, S., Lee, H., Park, S. et al. A comparison of determination methods for uranium radioactivity in environmental soil samples using a gamma spectrometer. J Radioanal Nucl Chem 331, 2929–2938 (2022). https://doi.org/10.1007/s10967-022-08379-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-022-08379-x