
Abstract
Radiolytic transformations of TODGA (N,N,N',N'-tetra-n-octylamide of diglycolic acid) in Isopar-M significantly depend both on the addition of alcohol (n-nonanol or n-decanol) and on the saturation of the solution with 8 M HNO3. Fragmentation of TODGA molecules is the dominant radiolytic process. Compared to acid-free solution, radiolysis in the presence of HNO3 results in a much lower yield of N,N-dioctylacetamide formation, while 2-hydroxy-N,N-dioctylacetamide remains the main degradation product of TODGA. The acidification of the solution promotes a lower radiolytic degradation of TODGA, but expands the range of final radiolysis products, in particular, due to the participation of NO2 and NO radicals in radiation-induced reactions. Alcohol serves as a source of alkoxy radicals, which are highly reactive towards TODGA. In the presence of nitric acid, a significant portion of alkoxy radicals decay in reactions with NO2, NO, and acyl radicals and, thus, do not participate in the decomposition of TODGA.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Nitric acid can significantly affect the mechanism of radiolysis of organic extraction systems used in radiochemical technology to extract radionuclides from HNO3 solutions [1]. During operation, the organic extractant solution is simultaneously irradiated and exposed to aqueous solutions of nitric acid of various concentrations, from 1 to 7 M. The high acidity of the nitric acid medium and a wide range of reactive nitrogen-containing radicals [2, 3] capable of trapping radiolytic intermediates arising from the extractant and solvent are the main factors that influence the behavior of the system. Therefore, the elucidation of the effect of nitric acid on the radiolysis mechanism and the stability of the extraction system is a fundamentally important task.
TODGA, N,N,N',N'-tetra-n-octylamide of diglycolic acid, is one of the promising ligands for the extraction of An(III) and Ln(III) from highly concentrated solutions of nitric acid [4,5,6,7,8]. Mixtures of heavy alkanes and aliphatic alcohol [5, 9], in particular solutions of n-nonanol or n-decanol in Isopar-M [10, 11], are suitable diluents for TODGA. Previously, we studied the composition of the radiolysis products of TODGA hydrocarbon-alcohol solutions in the absence of nitric acid [10]. It was shown that the dominant radiolytic transformation of excited molecules and radical cations of TODGA consists in the cleavage of the ether bond with the subsequent formation of N,N-dioctylacetamide and 2-hydroxy-N,N-dioctylacetamide. Also, an important role is played by the dissociative addition of •H, •OH and alkoxy radicals to the carbonyl group TODGA, leading to the cleavage of the C–N and C–C bonds in the α-position relative to the carbonyl group. This work is devoted to the study of the effect of extracted nitric acid on the radiolytic transformations of TODGA in mixtures of n-nonanol or n-decanol with Isopar-M.
Experimental methods and conditions
In order to adequately compare the radiolysis of unacidified and acidified solutions, irradiation, mixing of organic components, and analysis of the products were carried out under the same conditions as in [10].
Solutions and extraction mixtures
The investigated extraction mixtures are based on TODGA (N,N,N’,N’-tetra-n-octylamide of diglycolic acid) in the complex diluent Isopar-M (a mixture of isoparaffins with boiling range of 208–257 °C) with aliphatic monohydric alcohol (n-decanol or n-nonanol) provided by JSC “V.G. Khlopin Radium Institute”. In addition to Isopar-M, the S1 mixture contained 0.15 M TODGA and 6 vol% n-decanol, the S2 mixture—0.15 M TODGA and 6 vol% n-nonanol, the S3 mixture—0.2 M TODGA and 20 vol% n-decanol, the S4 mixture—0.2 M TODGA and 20 vol.% n-nonanol. Before irradiation, the solutions were saturated with nitric acid by stirring three times with an aqueous solution of 8 M HNO3 in a volume ratio of 1:1. The HNO3 content in the organic phase was determined by potentiometric titration on the Akvilon ATR-02 device with glass and calomel electrodes. Aliquots of HNO3-saturated solutions were diluted with a 1:3 mixture of acetone and ethanol and titrated with an alcoholic NaOH solution. The HNO3 concentration was 1.15 mol dm−3 in S1 and S3 samples, and 1.2 mol dm−3 in S2 and S4 samples.
A source of ionizing radiation and radiolysis of the extraction mixtures
The extraction mixtures were irradiated in a cylindrical glass reactor with a water seal at an ambient temperature of 17 ± 2 °C using the UELV-10-10-S-70 linear accelerator with a scanning electron beam (energy of 8 meV, pulse duration of 6 μs, pulse repetition rate of 300 Hz, average beam current of 700 μA, vertical scan frequency of 1 Hz, sweep width of 245 mm). Irradiation was carried out in an intermittently periodic mode: the irradiation interval up to a dose of 10 kGy (dose rate 0.22 kGy s−1) alternated with an interval of sample cooling for 10 min. Phenazine dye-doped copolymer film standard reference material SO PD(F)R-5/50 [GSO (Certified Reference Material) no. 7875–2000] was used as dosimeter. The total absorbed dose for each sample was 500 kGy.
IR and chromatographic analysis
The content of carboxylic acids, esters, nitro compounds and ketones in irradiated solutions was determined on IR Prestige-21 (Shimadzu) spectrophotometer using calibration curves for the corresponding functional groups: −COOR at 1740 cm−1 (butyric acid hexyl ester), −COOH at 1730 cm−1 (myristic acid), −NO2 at 1556 cm−1 (2-nitrooctane), and =C=O at 1721 cm−1 (4-methyl-2-pentanone). The light source was a JDU Uniphase helium–neon laser with a wavelength of 632.8 nm. The spectra were recorded using CaF2 glasses and a cuvette with a lead gasket 0.129 mm thick. The presence of ketones in the samples was proved by the qualitative reaction with 2,4-dinitrophenylhydrazine.
The component composition of the radiolysis products was determined after neutralizing the samples with a 1:1 mixture of sodium bicarbonate and anhydrous sodium sulfate. Neutralization was used to avoid acid and water damage to the chromatographic column and detector. Sodium bicarbonate served as a neutralizing agent and anhydrous sodium sulfate served as a water scavenger. The samples were analyzed using a Thermo Scientific Trace 1310 gas–liquid chromatograph with an ISQ mass-spectrometer detector (ionization by electrons, 70 eV) and a Trace 1310 gas–liquid chromatograph with a flame ionization detector. The samples were dissolved in acetone for analysis. The flow split mode was used (1:20 and 1:5). Helium was used as a carrier gas (flow rate 1.2 mL min−1). Thermo columns with a polydiphenylsiloxane/polydimethylsiloxane ratio equal to 5:95 of 15 m (TG-5MS, 15 m × 0.25 mm) and 30 m long (TG-5MS, 30 m × 0.25 mm) were used for qualitative and quantitative analysis, respectively. Products were identified by mass spectra and retention indices using the NIST-2017 database. According to the results of repeated experiments, the relative error of measurements did not exceed 10%.
Results
According to IR spectroscopic analysis, irradiated solutions, before neutralization, contain a wide range of products, including organic acids, esters, nitro compounds and ketones (Fig. 1). The presence of nitric acid promotes radiolytic formation of carboxylic acids and esters, especially in S3 and S4 solutions with higher alcohol content. Therefore, alcohol can be considered as an important precursor of carboxyl compounds. The acid also stimulates the radiolytic formation of ketones, but their yield is higher in S1 and S2 solutions with lower alcohol content. In turn, the radiolytic formation of nitro compounds is practically independent of the concentration of both alcohol and TODGA.

Content of carboxylic acids, esters, nitro compounds and ketones in irradiated S1-S4 solutions (before neutralization)
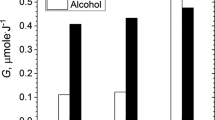
Judging by the composition of the products in neutralized solutions, acidification and irradiation make very different separate contributions to the decomposition of the dissolved components. For example, preliminary saturation of S1 and S2 solutions with nitric acid has relatively little effect on TODGA and alcohol (Fig. 2). Radiolysis of acid-free S1 and S2 solutions also weakly affects alcohol, but provides intense degradation of TODGA. The combined action of acid and ionizing radiation has a synergistic effect on alcohol degradation, but comparatively less effect on TODGA degradation. This response means that nitric acid somewhat slows down the reactions of radiolytic intermediates with TODGA, but significantly accelerates their reaction with alcohol. The synergistic effect of enhanced degradation is observed for both n-decanol (in S1) and n-nonanol (in S2).

Content of TODGA (a) and alcohol (b) in S1 and S2 solutions (after neutralization). Actions: + A means saturation with 8 M HNO3, + R means 0.5 MGy irradiation
In the presence of acid, the range of radiolysis products expands (Table 1), primarily due to the formation of organic nitrites, nitrates and nitrosamines (P12-P14). The content of P12 and P13 in irradiated solutions is much lower (radiation-chemical yield G is less than 0.05 μmol J−1) than P14. The dominant alkyl groups in nitrites and nitrates are n-decyl (in S1 and S3) and n-nonyl (in S2 and S4), which indicates the participation of the corresponding alcohols, decanol and nonanol, in the formation of P12 and P13. The main radiolytic product due to the presence of HNO3 is N-nitroso-di-n-octylamine (P14). It is formed in all S1-S4 solutions. Its yield depends on the alcohol content, but almost does not depend on the type of alcohol.
Nitric acid has a significant effect on the radiation-chemical yields of the final products (Fig. 3). In both S1 and S2, the most noticeable effect is a manifold decrease in the yield of N,N-dioctylacetamide (P1). In the absence of acid, P1 is the dominant product of the radiolytic degradation of TODGA, and in the HNO3-saturated solution it becomes one of the minor products. In turn, the yields of P2 are high in both acid-free and acidified solutions. The presence of nitric acid also contributes to a noticeable increase in the yields of P4, P6, and P7, while the yields of P3 and P5 are weakly dependent on the acidification of solutions.

Radiation-chemical yields G of the main degradation products of TODGA in S1 solution (after neutralization)
The increased alcohol content (in S3 and S4) leads to higher yields of P5, P6 and P14 in acidified solutions (Fig. 4). The yields of the P1-P4 products remain almost the same as in S1 and S2, but the yields of P7 are slightly reduced. The observed changes in yields show that the formation of P1 and P4 is most sensitive to the presence of acid, while the formation of P5 and P6 is more sensitive to the alcohol content.

Radiation-chemical yields G of the main degradation products of TODGA in S4 solution (after neutralization)
Acidification has a significant effect on the radiolytic formation of P10 esters (Fig. 5). Compared with acid-free solutions, the yield of P10 in acidified solutions increases several times. Moreover, the higher the alcohol content in the acidified solution, the higher the P10 yield (Fig. 5). The ether unit –O– in P10 is combined with an n-decyl group (in S1 and S3; y = 10) or with an n-nonyl group (in S2 and S4, y = 9), while the carbonyl group is combined with alkyl groups of various lengths (x = 3—9). There are two maxima in the x distribution: x = 8 corresponds to the n-octyl radical present in TODGA, and x = 6 corresponds to the n-hexyl radical resulting from the cleavage of some central C–C bond in the alkane molecule (Isopar-M).

Radiation-chemical yield G of P10 esters in S1-S4 solutions depending on the length of the alkyl group y (a) and in S2 solution depending on the length of the alkyl group x (b) after neutralization
Discussions
H-radicals formation and reactions
High absorbed dose and high dose rate of electron-beam irradiation provide a wide range of final products resulting from both the primary and secondary radiolytic processes. A direct consequence of the participation of HNO3 in radiolytic transformations is the formation of nitroso and nitro derivatives (P12-P14) via the combination of organic radicals with NO and NO2 radicals. As shown earlier [12, 13], in the presence of oxidative radical scavengers, such as TODGA, aliphatic alcohol, and Isopar-M, the dominant transformation of nitrate ions consists in sequential reduction according to a simplified scheme
Deeper reduction gives N2O and N2, which are not highly reactive. Organic radicals, including alkyl and hydroxyalkyl radicals characteristic of the considered radiolysis, are also capable of taking part in the reduction of nitrate [14,15,16,17,18,19], for example
At the later stages of reduction (1), the H radicals play the most important role, since free and solvated electrons have a much shorter lifetime. In addition to reactions with TODGA molecules, electrons rapidly decay in reactions with proton (k = 2.4 × 1010 dm3 mol−1 s−1) and nitrate ion (k = 9.6 × 109 dm3 mol−1 s−1) [12, 20]
A decrease in the degree of TODGA degradation in the presence of nitric acid (Fig. 2) indicates a lower reactivity of H radicals in comparison with solvated electrons in reactions with TODGA. In the absence of nitric acid, electrons are captured predominantly by TODGA molecules (dissociative capture), since alkanes and alcohol have low reactivity with an electron [21]. The acid suppresses the reactions of electrons with TODGA, but promotes reactions with the participation of H radicals. Organic radicals are partially decay via competitive reactions with intermediates of the process (1) and, as a result, react less with TODGA. In addition, the acidic environment favors the enolization of TODGA, which somewhat complicates the cleavage of the C–C bond in the α-position relative to the carbonyl group.
Thus, the presence of nitric acid provides an increase in the yield of H radicals by reaction (3) in addition to radiation-induced H radicals arising from alkanes, alcohol, and alkyl groups of TODGA [10]. H radicals are partially involved in the reduction process (1), while the rest are involved in the formation of alkyl radicals by the mechanism of H-abstraction from alkanes and alkyl groups or H-addition to double bonds of TODGA and unsaturated radiolytic products.
Processes involving alkoxy radicals
Another important function of excess H radicals is the formation of additional alkoxy radicals (R-O•) due to H-abstraction from the hydroxyl group of the alcohol. Among the organic radicals formed in the system under consideration, the most reactive are alkoxy radicals [22,23,24,25,26]. They are formed via the decomposition of excited molecules and radical cations of alcohol, as well as via the abstraction of the H radical from the hydroxyl group. Alkoxy radicals are capable of rapidly removing H atoms or adding to double bonds. Obviously, in the presence of nitric acid, alkoxy radicals can recombine with NO2 and NO radicals. This recombination results in P12 alkyl nitrites and P13 alkyl nitrates
As a result of irradiation, n-decyl nitrite and n-decyl nitrate appear in acidified S1 and S3 solutions, and n-nonyl nitrite and n-nonyl nitrate appear in S2 and S4 solutions. No other alkyl nitrates and alkyl nitrites were seen. On the one hand, this indicates that the precursors of alkyl nitrites and alkyl nitrates are precisely alkoxy radicals arising from the corresponding alcohol: n-decanol in S1 and S3, and n-nonanol in S2 and S4. On the other hand, it is NO and NO2 that are the main intermediates in the radiolytic transformations of nitric acid in the systems under consideration. The most convincing evidence for the important role of NO is the formation of N-nitroso-dioctylamine (Figs. 2 and 3). Most likely, it is formed via the recombination of NO with the bulky •N(C8)2 radical, which appears during the radiolytic fragmentation of TODGA. Under reducing conditions, radical nitrosation
Occurs quite often [27,28,29] and is a characteristic process in the radiolysis of organic compounds in the presence of nitric acid [14]. Most of the NO2 radicals decay via recombination with alkyl radicals to form nitroalkanes (Fig. 1).
Fragmentation of the TODGA molecule
The radiation-chemical yield of TODGA molecules decay is approximately two times lower than the total yield of TODGA degradation products. For example, the yield of TODGA fragments in acidified S2 solution is slightly more than 0.4 μmol J−1, while the observed degradation yield of TODGA molecules does not exceed 0.17 μmol J−1. The yield of TODGA degradation in S3 and S4 solutions reaches 0.22, while the yield of TODGA fragments increases to almost 0.65 μmol J−1. This indicates that fragmentation is the main radiolytic process for TODGA. Table 1 indicates that more than two TODGA fragments can participate in the formation of final radiolytic products.
The induction effect of O atoms of the carbonyl and ether groups weakens the C–C bonds between them and the C-N bond adjacent to the carbonyl group. The influence of O and N atoms on the radiolytic cleavage of adjacent bonds is characteristic of oxygen- and nitrogen-containing compounds [30, 31]. The structure of the TODGA fragmentation products indicates a high probability of cleavage of C–O ether bond, C–C bond at the carbonyl group, as well as C–N bonds. In S1-S4 solutions, the cleavage of the C–O bond is observed twice as often as the cleavage of the C–C bond. The cleavage products of the C–O ether bond are P1, P2, P9 and P17. The bond cleavage can be caused by the excited state of the molecule or radical [10]. The relatively high yield of P2 indicates that its precursor is an alkoxy type radical

which quickly turns into P2 due to the abstraction of H atom from neighboring molecules. The low yield of P1 indicates that its precursor is an alkyl type radical, which is characterized by slower recombination processes with alkyl radicals from Isopar-M or alcohol to form P9. There is a wide range of P9 homologues, but their diversity and overlap with other peaks in the chromatogram make quantification difficult. The radical center between the ether and carbonyl groups can arise due to H-abstraction from the methylene group or due to H-addition to the carbonyl group. Both cases are favorable for cleavage of the bond in the α- or β-position relative to the radical center. Judging by the structures of P1 and P2, they are formed predominantly during the decay of excited TODGA molecules or during the decomposition of the TODGA H-adduct.
The primary and secondary products of C–C bond cleavage at the carbonyl group are P3, P4, P8, P9, P10, P15 and P16. Probably, P3 and P4 arise as a result of the H-adduct decay.

Due to the acid-catalyzed enolization of TODGA, the structure of the H-adduct can be different and, accordingly, its decay can lead to different products. A higher yield of P4 indicates that it is formed in molecular form. The carbon-centered radical RC then recombines with alkyl radicals (in particular, giving P3) or decomposes, for example, with the elimination of CO. In turn, the RA radical via recombination with other radicals forms P9, P10, P15, and P16. At the same time, the P8 structure indicates the possibility of cleavage of the α-C–C bond as a result of the addition of an alkoxy radical to the carbonyl group. The resulting alkoxy adduct further decomposes into P8 and the RC radical.

However, the addition of an alkoxy radical to a carbonyl group more often leads to cleavage of the α-C-N bond.

In S3 and S4 solutions, containing more alcohol, the cleavage of the α-C-N bond occurs almost three times more often than in S1 and S2 solutions. This indicates the dominant role of alkoxy radicals in the cleavage of the α-C-N bond. The primary and secondary products of the α-C-N bond cleavage are P5 and P14, as well as the alkoxy adducts P6, P10, and P11. The P11 product is the result of the addition of alkoxy radicals to both carbonyl groups of the TODGA molecule with the cleavage of adjacent C-N bonds. The probability of such a reaction is lower than that of the (10) and (11) reactions, but the P11 product is reliably detected in irradiated solutions. About half of the RN radicals are involved in the formation of N-nitroso-di-n-octylamine P14. Cleavage of the C-N bond within the di-n-octylamine group is less frequent than cleavage of the α-C-N bond. The probability of elimination of the octyl group somewhat decreases in S3 and S4, i.e. with an increase in the content of alcohol, which is a more effective scavenger of H radicals. This indicates that H radicals play a significant role in the formation of P7.

For S1 and S3 solutions, the length of the alkyl radical (Cy) in P6 and P8 products is equal to 10 C atoms. In turn, in S2 and S4 solutions, the alkyl group (Cy) contains 9 C atoms. Consequently, it is the alkoxy radicals of decanol and nonanol that are involved in (10) and (11) reactions. The addition of alkoxy radicals to the carbonyl group can also manifest itself in a decrease in the observed yield of ketones in S3 and S4 compared to S1 and S2 solutions containing less alcohol (Fig. 1). As noted above, alkoxy radicals decay in several competitive processes. In particular, reactions (5), (6), (10) and the like compete with reaction (11). Therefore, the observed yield of P6 via reaction (11) in acidified S3 and S4 solutions is only slightly higher than in acid-free solutions, where reactions (5) and (6) are absent (Fig. 4). In acid-free S1 and S2 solutions (Fig. 3), containing less alcohol and alkoxy radicals, TODGA and its fragmentation products, including P6, decay mainly in reactions with a solvated electron. At a high concentration of alcohol, this effect is weakened due to the scavenging of solvated electrons by hydroxyalkyl radicals, carbonyl products, and alcohol.
Formation of secondary products
The high yield of H radicals gives them the opportunity to participate in secondary processes. In particular, the addition of the H radical to the carbonyl group (or the enol C = C bond) of the P6 molecule can lead to the cleavage of the α- or β-C–C bond with the formation of P2 or P3, as well as the RE1 or RE2 carbon-centered radical.

Further, RE1 and RE2 radicals participate predominantly in recombination with alkyl radicals, forming P10 esters, where x = 3–10 (Fig. 5). Most likely, (13) reaction leads to the RE2 radical. In turn, the dimerization of RE1 radicals results in the P17 product formation.
The predominance of alkyl groups in the components of the solution provides the highest probability of radiolytic formation of alkyl radicals. However, low-reactive alkyl radicals, apparently, cannot compete with alkoxy radicals in reactions with NO and NO2. As a consequence, the products of the recombination of alkyl radicals with NO or NO2 are hardly noticeable in irradiated solutions. Both alkanes and alcohols can undergo radiolytic cleavage of one of the internal C–C bonds. As a rule, alkyl chains are cleaved at the bond located in the β-position relative to the radical center. The relative probabilities of radiolytic cleavage of C-H and C–C bonds in alkyl groups are approximately 2:1 [14]. Fragmentation of alkane (Isopar-M) and alcohol molecules via cleavage of the C–C bond leads to the appearance of short alkyl and hydroxyalkyl radicals with the subsequent formation of the corresponding low molecular weight hydrocarbons, aldehydes and alcohols. Alkanes and alcohols can serve as precursors of unsaturated compounds. Excited alkane and alcohol molecules can decay via eliminating a hydrogen molecule and forming alkenes and aldehydes, respectively. Similar alkenes and aldehydes arise from disproportionation of alkyl (•CnH2n-1) or hydroxyalkyl (•CxH2xOH) radicals
The carbonyl group of an aldehyde, like TODGA, is a radical scavenger. At the same time, the acidic environment promotes the isomerization of aldehydes into the enol form. Due to the dynamic interconversion of C-H and O–H bonds, i.e. mobility of a hydrogen atom, the reaction of radicals with enol can occur by the mechanism of dissociative addition [32, 33]

A similar dissociative addition of an oxidizing radical to an aldehyde with the formation of a carboxylic acid ester was observed earlier [34]. Reaction (17) serves as an additional source of free H radicals. In addition, in the considered extraction system, it causes additional formation of P10 esters (Fig. 5). Decyl esters predominate in S1 and S3, while nonyl esters prevail in S2 and S4, which is evidence of the participation of intermediates of the corresponding alcohol in the formation of esters. In turn, the addition of an H radical to an aldehyde can be a secondary source of alkoxy radicals.

Radiolytic processes involving TODGA adducts such as P6, P8, P9, P11, and P15 can serve as an alternative source for the formation of P10 esters. Another way of P10 formation can be due to ion-molecular reactions. The interaction of alkane or alcohol radical cation with an aldehyde can form an alkanoyl (acyl) radical.

A similar radical occurs when H is abstracted from the aldehyde by small radicals. Acyl radicals can recombine with each other or with alkoxy radicals. In the first case, P16 oxalic acid esters appear. In the second case, P10 ester is formed. Aldehydes are also precursors of carboxylic acids observed in irradiated solutions prior to neutralization (Fig. 1). A carboxylic acid can be formed as a result of reactions of an acyl radical with water, an OH radical or an alkoxy radical (dissociative addition), as well as as a result of fragmentation of esters.
Conclusion
Saturation of the extraction system with 8 M HNO3 leads, on the one hand, to a decrease in the yield of TODGA degradation and, on the other hand, to an expansion of the range of final radiolytic products. The acid suppresses the reactions of dissociative addition of an electron to TODGA due to the fast conversion of electrons into less reactive H radicals. In addition, a significant portion of electrons are spent on nitrate reduction. Some of the formed NO2 and NO radicals combine to alkoxy radicals to form the corresponding organic nitrites and nitrates. Thus, acidification of the solution causes a decrease in the yield and average reactivity of radicals that could participate in the degradation of TODGA.
Under acidification conditions, an increase in the yield of H atoms results in an increase in the yield of alkyl radicals from Isopar-M and alcohol, as well as alkoxy radicals from alcohol. This effect is especially noticeable in S3 and S4 systems with high alcohol content. Thus, in acidified solutions, the formation of products of recombination of alkyl, hydroxyalkyl, and alkoxy radicals with each other and with long-lived TODGA radicals is intensified. In particular, these conditions favor the formation of carboxylic acid alkyl esters. The high yield of alkyl radicals and the corresponding intensification of their reactions are also the main reason for a significant decrease in the yield of N,N-dioctylacetamide (P1) in acidified solutions compared to acid-free solutions. Probably, the main precursor of P1 is the C-centered radical, which readily recombines with alkyl radicals. As a consequence, the formation of P1 is suppressed, while the formation of P9 becomes more important.
The yield of formation of TODGA fragments is more than twice the observed yield of degradation of the TODGA molecules themselves. This confirms that both in acid-free and acidified solutions, fragmentation of the TODGA molecule is the main radiolytic process. Moreover, the cleavage of the ether C-O bond occurs mainly during the decay of excited molecules and ions, while the cleavage of the C–C bond in the α-position relative to the carbonyl group occurs mainly as a result of the addition of radicals to the carbonyl group. The H and alkoxy radicals play the main role in the cleavage of the C–C bond, and the role of alkoxy radicals increases with an increase in the alcohol content in the solution.
References
Mincher BJ (2015) Radiation chemistry in the reprocessing and recycling of spent nuclear fuels. In: Reprocessing and recycling of spent nuclear fuel. Elsevier
Liu Z, Fang Z, Wang L, He H, Lin M-Z (2017) Alpha radiolysis of nitric acid aqueous solution irradiated by 238Pu source. Nucl Sci Tech. https://doi.org/10.1007/s41365-017-0200-4
Horne GP, Donoclift TA, Sims HE, Orr RM, Pimblott SM (2016) Multi-scale modeling of the gamma radiolysis of nitrate solutions. J Phys Chem B. https://doi.org/10.1021/acs.jpcb.6b06862
Iqbal M, Huskens J, Verboom W, Sypula M, Modolo G (2010) Synthesis and Am/Eu extraction of novel TODGA derivatives. Supramol Chem. https://doi.org/10.1080/10610278.2010.506553
Whittaker D, Geist A, Modolo G, Taylor R, Sarsfield M, Wilden A (2018) Applications of diglycolamide based solvent extraction processes in spent nuclear fuel reprocessing, part 1: TODGA. Solvent Extr Ion Exch. https://doi.org/10.1080/07366299.2018.1464269
Wilden A, Modolo G, Sypula M, Geist A, Magnusson D (2012) The recovery of An(Iii) in an innovative-sanex process using a todga-based solvent and selective stripping with a hydrophilic BTP. Procedia Chem. https://doi.org/10.1016/j.proche.2012.10.065
Lewis FW, Harwood LM, Hudson MJ, Geist A, Kozhevnikov VN, Distler P, John J (2015) Hydrophilic sulfonated bis-1,2,4-triazine ligands are highly effective reagents for separating actinides(iii) from lanthanides(iii) via selective formation of aqueous actinide complexes. Chem Sci. https://doi.org/10.1039/C5SC01328C
Jose J, Prathibha T, Karthikeyan NS, Venkatesan KA, Selvan BR, Seshadri H, Venkatachalapathy B, Ravichandran C (2020) Evaluation of selected solvent systems for the single-cycle separation of Am(III) from Eu(III) using aqueous soluble sulphonated bis-triazinylpyridine. J Mol Liq. https://doi.org/10.1016/j.molliq.2020.112893
Swami KR, Venkatesan KA, Antony MP (2019) Role of phase modifiers in controlling the third-phase formation during the solvent extraction of trivalent actinides. Solvent Extr Ion Exch. https://doi.org/10.1080/07366299.2019.1695560
Nikitina YV, Yudin NV, Belova EV, Ponomarev AV (2020) The effect of aliphatic alcohol additives on the radiolytic degradation of TODGA in Isopar-M. J Radioanal Nucl Chem. https://doi.org/10.1007/s10967-020-07375-3
Skvortsov IV, Belova EV, Yudintsev SV (2020) Effect of irradiation on the oxidation kinetics of TODGA-based extraction mixtures at atmospheric pressure. Nucl Eng Technol. https://doi.org/10.1016/j.net.2020.02.024
Bludenko AV, Makarov IE, Ponomarev AV (2002) Effect of dissolved oxygen on the radiolytic conversion of nitrate ions in aqueous solutions in the presence of formate. Mendeleev Commun. https://doi.org/10.1070/MC2002v012n04ABEH001594
Haas FM, Dryer FL (2015) Rate coefficient determinations for H + NO2 → OH + NO from high pressure flow reactor measurements. J Phys Chem A. https://doi.org/10.1021/acs.jpca.5b01231
Woods R, Pikaev A (1994) Applied radiation chemistry. Radiation Processing. Wiley, New York
Matsugi A, Shiina H (2017) thermal decomposition of nitromethane and reaction between CH3 and NO2. J Phys Chem A. https://doi.org/10.1021/acs.jpca.7b03715
Kroupnov AA, Pogosbekian MJ (2018) DFT calculation-based study of the mechanism for CO2 formation in the interaction of CO and NO2 molecules. Chem Phys Lett. https://doi.org/10.1016/j.cplett.2018.08.077
Rissanen MP, Arppe SL, Eskola AJ, Tammi MM, Timonen RS (2010) Kinetics of the R + NO2 Reactions (R = i–C3H7, n -C3H7, s -C4H9, and t -C4H9) in the Temperature Range 201–489 K. J Phys Chem A. https://doi.org/10.1021/jp909396v
Zhang J, Li Z, Liu J, Sun C-C (2006) Theoretical mechanistic study on the radical−molecule reaction of CH2OH with NO2. J Phys Chem A. https://doi.org/10.1021/jp055515x
Chai J, Goldsmith CF (2017) Rate coefficients for fuel + NO2: Predictive kinetics for HONO and HNO2 formation. Proc Combust Inst. https://doi.org/10.1016/j.proci.2016.06.133
Shin H-S, Kim Y-R, Ponomarev AV (2001) Influence of sulfite on radiolytic conversion of nitrate and nitrite in dilute aqueous solutions. Mendeleev Commun. https://doi.org/10.1070/MC2001v011n01ABEH001403
Ponomarev AV, Ershov BG (2020) The green method in water management: electron beam treatment. Environ Sci Technol. https://doi.org/10.1021/acs.est.0c00545
Dibble TS, Chai J (2017) Critical Review of atmospheric chemistry of alkoxy radicals. In: Advances in atmospheric chemistry. World Scientific
Guo J-J, Hu A, Zuo Z (2018) Photocatalytic alkoxy radical-mediated transformations. Tetrahedron Lett. https://doi.org/10.1016/j.tetlet.2018.04.060
Murakami M, Ishida N (2017) β-Scission of alkoxy radicals in synthetic transformations. Chem Lett. https://doi.org/10.1246/cl.170834
Ponomarev AV (2019) Acceleration of radical exchange and combination in 1-propanol under irradiation and boiling. Radiat Phys Chem. https://doi.org/10.1016/j.radphyschem.2019.01.011
Ponomarev AV, Kholodkova EM (2018) Boiling-induced changes in the mechanism of diglyme radiolysis. Mendeleev Commun. https://doi.org/10.1016/j.mencom.2018.07.011
Mullen C, Smith MA (2005) Low temperature NH(X 3Σ- ) radical reactions with NO, saturated, and unsaturated hydrocarbons studied in a pulsed supersonic laval nozzle flow reactor between 53 and 188 K. J Phys Chem A. https://doi.org/10.1021/jp045541f
Lazarou YG, Kambanis KG, Papagiannakopoulos P (1994) Gas-phase reactions of (CH3)2N radicals with NO and NO2. J Phys Chem. https://doi.org/10.1021/j100059a022
Berho F, Lesclaux R (2001) Gas phase reactivity of the cyclohexadienyl radical with O2 and NO and thermochemistry of the association reaction with NO. Phys Chem Chem Phys. https://doi.org/10.1039/b009136g
Kim Y, Ponomarev AV (2020) Radiolytic degradation of explosives in aqueous solutions and ‘red’ wastewater. Mendeleev Commun. https://doi.org/10.1016/j.mencom.2020.07.043
Ponomarev AV, Vlasov SI, Kholodkova EM, Chulkov VN, Bludenko AV (2019) Influence of boiling on radiolysis of oxygen-containing liquids. Radiat Phys Chem. https://doi.org/10.1016/j.radphyschem.2019.108405
Curran HJ (2006) Rate constant estimation for C1 to C4 alkyl and alkoxyl radical decomposition. Int J Chem Kinet. https://doi.org/10.1002/kin.20153
Ratliff BJ, Alligood BW, Butler LJ, Lee S-H, Lin JJ-M (2011) Product Branching from the CH2CH2OH Radical Intermediate of the OH + Ethene Reaction. J Phys Chem A. https://doi.org/10.1021/jp203127k
Cameron M, Sivakumaran V, Dillon TJ, Crowley JN (2002) Reaction between OH and CH3CHO. Phys Chem Chem Phys. https://doi.org/10.1039/b202586h
Acknowledgements
This work was financially supported by Ministry of Science and Higher Education of Russian Federation (topic № AAAA-A18-118021990023-6)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Informed consent
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Serenko, Y.V., Ponomarev, A.V., Belova, E.V. et al. Contribution of nitric acid and alcohol to the radiolytic degradation of TODGA in Isopar-M. J Radioanal Nucl Chem 328, 1319–1328 (2021). https://doi.org/10.1007/s10967-021-07732-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-021-07732-w