
Abstract
A novel method has been developed for the simultaneous determination of uranium, plutonium and americium nuclides in soil and sediment samples up to 5 g. Samples are destroyed by fusion with sodium hydroxide. Pre-concentration procedure tailored to the extraction chromatography is based on co-precipitation of actinides and removal of silica, iron and calcium. For the sequential separation of actinides a single DGA resin® (containing N,N,N′,N′-tetra-n-octyldiglycol-amide) column is used. Separation of americium from lanthanides is inherently involved in the EC procedure. Alpha sources are prepared from the individual actinides strip solutions. High recoveries (above 75%) and sensitivities (about 0.1 Bq/kg) have been obtained to allow the determination of actinides of environmental levels. The whole procedure can be performed in 2 days.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
A great variety of procedures for the selective separation of major and minor actinides (Pu, U, Th, Np, Am and Cm) and their determination by α spectrometry (AS) or mass spectrometry (MS) have been described and compiled in the excellent book of Lehto and Hou [1]. Ion exchange chromatography, liquid–liquid extraction and extraction chromatography (EC) have been successfully applied for the separation of single actinides or groups of them, but there are a few methods for the combined determination of all actinides. Since a great variety of specific resins became commercially available, e.g., by EiChrom Technologies [2] and TRISKEM International [3], coupling of chromatographic columns became a standard tool for developing combined procedures. A more economic and simpler solution is offered by the use of a single resin of sufficiently high selectivity where all actinides are retained on the resin followed by their sequential elution. Tetravalent actinides (Th, Pu, Np and U after oxidation state adjustment) are specifically retained by anion exchangers, TEVA (aliphatic quaternary amine), TK200 (trioctylamine), UTEVA (dipentylpentyl phosphonate), TRU (carbamoylmethyl phosphine oxide derivative) or DGA (N,N,N′,N′-tetraoctyldiglycolamide) resins. Tetra- and hexavalent actinides (Th, Pu, Np and U) are specifically retained by UTEVA, TRU and DGA resins. For the selective retention of trivalent actinides (Am, Cm, Pu after reduction) two resins are potentially applicable, TRU and DGA, but DGA is superior to TRU regarding the significantly higher capacity factors (k′) at higher acid concentrations. The highest k′ for Am retention on TRU resin is about 100 in 2 M HNO3 solution and it is reduced in the presence of Fe(III), while the maximum k′ for Am on DGA resin is more than 1000 when acidity is higher than 4 M HNO3 or 4 M HCl and it is not reduced by Fe(III). TRU and DGA resins can strongly retain tetra- and hexavalent actinides, as well.
A rapid method for the determination of all actinides using a single TRU resin column was developed and tested at IAEA’s Laboratories at Seibersdorf [4, 5]. It became one of the recommended methods of IAEA for emergency situation, but the method fails when bigger amounts (> 1 g) of soil or sediment are treated. Horwitz et al. proposed a method for the combined determination of U, Pu and Am using a single DGA column [6]. The sample was loaded on a 2 mL DGA column from 3 M HNO3 containing NaNO2 to stabilize Pu(IV) oxidation state. Uranium, Am and Pu were stripped consecutively with 0.1 M HNO3, 0.1 M HCl and 0.1 M ammonium oxalate. NaNO2 was added continuously also to the U and Am strip solutions probably to avoid Pu leaking. Recoveries of U, Am and Pu were as high as 82%, 97% and 95%, respectively. Unfortunately, in this procedure Th and Np can contaminate other actinide fractions [7].
According to the literature, DGA resin has been applied successfully for the separation and purification of Am and for the pre-concentration of actinides from various sample types. Maxwell et al. [8] included the DGA resin in the combined separation method of actinides after TEVA and TRU resins where Pu and Th were retained on the first resin, U and Am on the second one, and Am was collected finally on DGA. Luisier et al. [9] separated Pu and Am from water and soil samples using TEVA and DGA resins. Eickenberg et al. [10] concentrated actinides from soil sample on DGA, and used anion exchange resin for Pu, UTEVA for U and Th, and DGA for Am (and lanthanides) separation.
Recently, a procedure for the complex separation of all actinides has been developed at our laboratory [7]. Actinides were retained from 4 M HCl under reducing conditions on a small DGA column (0.5 g). Uranium, Th–Np, Pu and Am–Cm were eluted sequentially after on-column oxidation state adjustments with diluted acids, using complexing agent and changing the temperature of the column and the eluent. Wash fractions were included in the procedure between the eluates to reduce cross-contamination. In the chromatographic separation high recoveries (above 85%,) were obtained without significant cross-contamination (< 5%). The method was applied for the analysis of radioactive waste samples up to 100 mL. Samples were digested by evaporation with mineral acids in open system. Actinides were pre-concentrated with ferrous hydroxide, then chromatographically separated on the DGA column. Alpha sources were prepared by micro-co-precipitation from the strip solutions directly. High recoveries (above 50% for all actinides) and decontamination factors were obtained in the whole procedure.
The aim of the present work was to develop a fast and accurate procedure for the analysis of U, Pu, Am nuclides in environmental samples, especially soil and sediment in order to meet the requirements of monitoring the environment. To reach the necessary sensitivity for analysis of actinides of environmental levels minimum 5 g of samples have to be processed. Two types of sample destruction techniques, i.e. (1) acid destruction with microvawe digestion followed by evaporation of mineral acids in open system, and (2) fusion with NaOH according to the method of Maxwell et al. [8] were tested, comparatively evaluated. Fusion was favored for its rapidity and complete decomposition of even refractory particles. For rapid separation of actinides from the matrix and each other the chromatography with single DGA column as described above was tested. Two approaches were followed, the sample destruction procedures (1) and (2) were combined directly with the chromatographic separation. Both methods were tested with a set of standard reference materials. Unfortunately, both methods failed with certain sample types. Additionally, significant amount of gelatinous precipitate (silica gel) was formed when the fused samples were acidified what slowed down or occasionally blocked the chromatography.
To find out the reasons of failure, the elemental composition of the samples was measured and compared. Differences in the concentration of the major components (Fe, Ca, Al content) and some minor components of possible interference (Zr) were blamed for losses. The effect of these components on the retention properties of actinides was tested by batch experiments, and the k′ values of actinides were determined. It was found that the presence of high concentrations of Fe and Ca altered the retention of all actinides but in different ways. In order to adjust the Fe and Ca concentration in the load solution to the optimal range and to remove silica, actinide pre-concentration steps were included in the procedure between sample destruction and chromatography. Development of the process of pre-concentration is discussed in the present paper. Additionally, the separation of lanthanides from americium by chromatography on the same DGA column has been studied. By the removal of some lanthanides the resolution of americium alpha sources has been significantly improved. Finally the combined method has been tested with a set of standard reference materials.
Experimental
Batch uptake experiments
50–100 mg of DGA resin (N,N,N′,N′-tetra-n-octyldiglycolamide) (50–100 µm particle size) were contacted with 1.5 mL of a 4 M HCl solution spiked with known activities (10–50 Bq) of the radiotracers, i.e. 241Am, 239Pu, 230Th and 233U in PE centrifuge tubes. Solutions contained various amounts of reagents, i.e. Na2SO3 (0–62,500 ppm), FeCl3 (0–4375 ppm Fe), CaCl2 (0–12,500 ppm Ca), AlCl3 (0–6250 ppm Al), ZrOCl2 (0–5000 ppb Zr).
Samples were shaken for 60 min in a shaker, then settled for 10 min and the phases were separated with a 0.45 µm pore size Nylon syringe filter. Experiments were performed at room temperature (not controlled). The separated liquid phase was counted against blank and standard samples in a liquid scintillation counter. The capacity factor k′ was calculated using the following equation:
where A0 and As are the aqueous phase activity before and after equilibration, w is the weight of the resin in grams, and V is the volume of the aqueous phase in mL (see in Ref. [7]).
Tests to co-precipitate actinides with sub-stoichiometric amounts of Ca fluoride and Ca hydroxide in presence of Mg hydroxide
CaF2 test A Ca stock solution was prepared by dissolving 5.55 g Ca(OH)2 in 200 mL 1 M HCl, 7 mL 40% HF were added to form precipitate, then the precipitate was dissolved with 4 g H3BO3. 10 mL aliquot of this stock solution was spiked with one of the tracers 239Pu, 241Am, 233U or 230Th of about 10 Bq each. 0.1 g Fe(NO3)3·9H2O and 0.1 mL 70% hydrazine were added. The solution was warmed and 40% HF was added in various amounts between 100 and 1000 uL. Samples were centrifuged and 1 mL aliquot of the supernate was mixed with 15 mL ProSafe TS cocktail and counted in LSC against blank and standard prepared from the tracer. The % of activity in the sample against the initial activity was calculated. The amount of Ca in the CaF2 precipitate was determined by measuring the supernates using AAS.
Mg(OH)2 test A Mg stock solution was prepared by dissolving 3.02 g MgO in 200 mL 1 M HCl, 7 mL 40% HF and a few mL 25% NH3 were added to form precipitate, then the precipitate was dissolved with 4 g H3BO3 and a few mL 37% HCl. 10 mL aliquot of this stock solution was spiked with one of the tracers 239Pu, 241Am, 233U or 230Th of about 10 Bq each. 0.1 g Fe(NO3)3·9H2O and 0.1 mL 70% hydrazine were added. The solution was warmed and 5 M NaOH was added in various amounts between 100 and 1500 uL. The pH was measured. Samples were centrifuged and 1 mL aliquot of the supernate was mixed with 15 mL ProSafe TS cocktail and counted in LSC against blank and standard prepared from the tracer. The % of activity in the sample against the initial activity was calculated.
Ca(OH)2 test The test was performed with the Ca stock solution as described above for Mg(OH)2 test. Spikes were not added to the samples, LSC measurements were not performed. The amount of Ca in the Ca(OH)2 precipitate was determined by measuring the supernate using AAS.
Mixed Ca(OH)2/Mg(OH)2 test The experiments were performed with the Ca stock solution, additionally 84 mg of MgCl2 was added to each test sample. The test was performed as described above for Mg(OH)2 test. Spikes were not added to the samples, LSC measurements were not performed. The amount of Ca and Mg in the mixed Ca(OH)2/Mg(OH)2 precipitate was determined by measuring the supernate using AAS.
Chromatographic separation of actinides on DGA column
0.5 g DGA resin® (registered trade mark of TRISKEM International) of 50–100 μm particle size was soaked in 4 M HCl for a couple of hours, it was packed into PE chromatographic column with 7 mm inner diameter. Quartz sand was used for the top bed support against clogging. The column of DGA resin was preconditioned with 10 bed volumes of 4 M HCl solution. For temperature control jacketed columns were prepared by surrounding the standard PE columns with home-made glass jackets and circulating water from a standard temperature control unit. The temperature of the eluents was also controlled using the water bath system of the same unit (Fig. 1).

Photo of the chromatographic separation system with temperature control unit and vacuum box
The typical flow rate provided by a vacuum box joining to the bottom of the columns was 1 mL/min. The load/elution sequence is summarized in Table 1.
In course of R&D, steps 10 and 11 have been modified:
Step 10: Pu rinse, light lanthanide strip: 10 mL 0.5 M HNO3 + 3 mL 0.05 M HNO3 at 40 °C.
Step 11: Am strip: 5 mL 1.5 M HCl at 40 °C.
Procedures for determination of actinides in soil and sediment
Procedure I: acid destruction and chromatography on DGA column
The procedure consists of sample destruction by evaporation with mineral acids (a), oxidation state adjustment of actinides and load preparation (b), chromatographic separation of actinides (c), α source preparation (d) (see the flowchart in Fig. 3a).
-
(a)
Sample destruction by evaporation with mineral acids
Samples of about 4 g are destroyed with HNO3, HCl and HF using a microwave (MW) oven followed by evaporation in open system. The dry sample (not ashed) is weighted in 8 Teflon microwave tubes. 7 mL 65% HNO3, 2 mL 37% HCl and 2 mL 40% HF are added to each tube and destruction is performed in an Anton Parr MW3000 facility. The samples are combined and placed in a Teflon beaker, spikes are added. The destruction is followed by evaporation consecutively with additional 40% HF, 65% HNO3 and 37% HCl of about 20–50 mL each depending on sample composition. The residue is taken up with 50 mL 0.5 M HCl and 2–4 g H3BO3. The solution is filtered through a cellulose nitrate membrane of 0.45 μm pore size and 40 mm diameter.
-
(b)
Oxidation state adjustment of actinides and load preparation
1–2 g of Na2SO3 are added to the solution to reduce actinides. The solution is warmed and stirred for 10 min and the reduction of Fe is checked by a negative thiocyanate test. The solution is cooled to room temperature in water bath and 25 mL 37% HCl is added to adjust acidity to about 4 M.
-
(c–d)
Chromatographic separation of actinides from the 80 mL load and α source preparation are performed in the same way as in Procedure III.
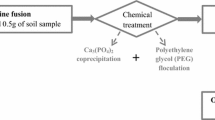
Procedure II: Alkaline fusion and chromatography on DGA column
The procedure consists of sample destruction by fusion (a), oxidation state adjustment of actinides and load preparation (b), chromatographic separation of actinides (c), α source preparation (d) (see the flowchart in Fig. 3b).
-
(a)
Sample destruction by fusion with NaOH, dissolution of the melt
Destruction of about 5 g of sample (+ spikes) with 30 g of NaOH in zirconium crucible is performed similarly than in Procedure III. The melt is dissolved with 100 mL distilled water and transferred to a glass beaker. The suspension is carefully acidified with 37% HCl to about 0.5 M acidity. A gelatinous suspension is obtained that is centrifuged and the supernate is filtered through a cellulose nitrate membrane of 0.45 μm pore size and 40 mm diameter. If the filter is blocked by the sample even after centrifuging than 1–2 g of polyethyleneglycol (PEG) is added to coagulate silica gel and help filtration.
-
(b)
Oxidation state adjustment of actinides and load preparation
3 g of Na2SO3 are added to the solution to reduce actinides. The solution is warmed and stirred for 10 min and the reduction of Fe is checked by a negative thiocyanate test. The solution is cooled to room temperature in water bath and 100 mL 37% HCl is added to adjust acidity to about 4 M. The solution is centrifuged and the supernate is filtered again without or with help of 1–2 g of PEG through cellulose nitrate membrane of 0.45 μm pore size and 25 mm diameter.
-
(c–d)
Chromatographic separation of actinides from the 300 mL load and α source preparation are performed in the same way as in Procedure III.
Procedure III: alkaline fusion, pre-concentration of actinides and chromatography on DGA column
The procedure consists of sample destruction by fusion (a), actinides co-precipitations using mixed (ferrous) hydroxides (b), mixed (calcium) fluorides (b) and mixed (magnesium) hydroxides (c), oxidation state adjustment of actinides and load preparation (d), chromatographic separation of actinides (e), α source preparation (f) (see the flowchart in Fig. 5).
-
(a)
Sample destruction by fusion with NaOH, dissolution of the melt
5 g of dry soil, sediment is weighted in a 250 mL zirconium crucible. Spikes are added in small droplets on the top of the sample and the liquid is evaporated under IR lamp. Typically 0.05–0.10 Bq of the following spikes are used: 232U, 242Pu, 243Am. 30 g of NaOH flakes are layered on the top and the crucible is placed in an ashing oven. The temperature is raised slowly (during about 1 h) up to 550 °C. At this temperature the fusion takes 1 h. Then the crucible is taken from the oven and the sample is cooled.
The solidified melt is dissolved with five 150 mL portions of distilled water that are transferred to 1 L glass beaker. The slurry is heated to boiling on a hot plate while stirring intensively. The zirconium crucible is rinsed twice with 5 mL 37% HCl that is heated to boiling under a glass watch and the solution is added to the NaOH solution.
-
(b)
Actinides co-precipitation with mixed (ferrous) hydroxides, dissolution of the mixed hydroxides
5 mL of 40% hydrazine and 1 g of Mohr’s salt (Fe(NH4)2(SO4)2·6H2O) dissolved in 20 mL water are added to the solution to obtain in situ ferrous hydroxide precipitate that can co-precipitate all actinides including uranium. The solution is boiled and stirred for an hour, then cooled in water bath to let the precipitate settle. The precipitate is filtered through a polyethersulfone membrane of 0.45 μm pore size and 40 mm diameter. Filtration of the well settled precipitate takes less than half an hour. The precipitate is washed with 100 mL 0.1 M NaOH.
The precipitate is transferred from the filter funnel into the original beaker with about 30 mL 67% HCl that is evaporated to dryness. The residue is taken up with 100 mL 1 M HCl. It is heated and stirred.
-
(c)
Actinides co-precipitation using mixed fluorides, dissolution of the mixed fluorides
About 2.5 mL 70% hydrazine are added to reduce iron. If necessary further 1 mL portions of hydrazine and 25% NH3 are added to complete the reduction of iron indicated by a negative Fe(III)-SCN test. The solution (or gelatinous slurry) is transferred to a 200 mL Teflon beaker and 25 mL 40% HF are added to the hot solution that is stirred for half an hour, then cooled in water bath. Fluoride precipitates are filtered through a cellulose nitrate membrane of 0.45 μm pore size and 25 mm diameter using plastic funnel. Filtration that removes silica remains is fast (typically 10 min).
The precipitate is transferred with 30 mL 3 M HNO3 to a glass beaker that contains 2 g of boric acid. The sample is heated to boiling for 10 min. Additional amount of boric acid (up to 1 g) and a few mL of 65% HNO3 may be added if the sample does not seem to be dissolved. The solution that might be slightly opaque is diluted with distilled water to 100 mL.
-
d)
Actinides co-precipitation using mixed (magnesium) hydroxides, dissolution of the mixed hydroxides
1 g of Fe(NO3)3·9H2O and 1 mL 70% hydrazine are added to the hot solution that is heated and stirred as long as Fe(III) is reduced to Fe(II) as indicated by a negative thiocyanate test. If it is needed few more drops of hydrazine can be added. 7.5 g of MgCl2·6H2O are added and dissolved. The pH of the solution is adjusted to 6–7 by the addition of 5 M NaOH (about 15 mL) when greenish-brownish colored precipitate is formed. The suspension is warmed and stirred for further 15 min, then cooled to room temperature in water bath, centrifuged and filtered through cellulose nitrate membrane of 0.45 μm pore size and 25 mm diameter.
The precipitates are combined and transferred with 20–30 mL 37% HCl to a glass beaker. The solution is evaporated to dryness, evaporation is repeated with 2 × 5 mL 65% HNO3. The residue is dissolved in 5–10 mL 37% HCl and evaporated again to dryness, finally taken up with 40 mL 0.5 M HCl.
-
(e)
Oxidation state adjustment of actinides and load preparation
1 g of Na2SO3 and 0.5 g of Mohr’s salt are added to the solution to reduce actinides. The solution is warmed and stirred for 10 min and the reduction of Fe is checked by a negative thiocyanate test. (The solution is not necessarily clear during this step). The solution is cooled to room temperature in water bath and 20 mL 37% HCl are added to adjust acidity to about 4 M. The solution is filtered through cellulose nitrate membrane of 0.45 μm pore size and 25 mm diameter. (If filtration is slow centrifuging may precede the filtration.)
-
(f)
Chromatographic separation of actinides on DGA column
The chromatographic separation of the 60 mL load is performed as described in Experimental Sect. 2.3.
-
(g)
α source preparation
Micro-co-precipitation is used for α source preparation as follows:
A solution containing 50 μg Nd as Nd(NO3)3 and 5 mL 40% HF are added to the strip solutions of uranium, thorium, americium. To the uranium strip solution 400 mg Mohr’s salt are added, too. In case of plutonium, the strip solution is first evaporated, persulfate and oxalate ions are decomposed by evaporation with 3x2 mL 65% HNO3. The residue is taken up with 20 mL 0.5 M HNO3. Then Mohr’s salt, Nd solution and HF are added as in case of the uranium source. After half an hour, micro-precipitates are filtered slowly through a membrane (e.g., polyethersulfone) of 0.10 μm pore size and 25 mm diameter. Filters are dried under IR lamp.
Tests to separate lanthanides from americium
An elution test was performed with the following model solution: 10 mL of a mixed standard solution containing 0.1 μg of each of the following elements La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu (totaling 1.4 μg) was acidified with 5 mL 37% HCl. The solution was loaded on the standard DGA resin column (0.5 g resin, column inner diameter 7 mm), and the separation scheme described in the Experimental Sect. 2.3 was followed till the elution of Pu. Then the following eluents were used: 10 mL 0.5 M HNO3, 3 mL 0.05 M HNO3, 5 mL 1.5 M HCl. Each mL of the eluates was collected separately and analyzed after dilution for lanthanides by ICP-MS.
5 g of IAEA-300 sediment sample were processed by Procedure III described in Experimental Sect. 2.4.3. 100 mL load solution was passed through the standard DGA column, and the chromatography was performed as described in Experimental Sect. 2.3. till stripping Pu. Then the column was eluted with 10 mL 0.5 M HNO3, 3 mL 0.05 M HNO3 and 5 mL 1.5 M HCl. Lanthanides in each mL of the eluates were measured by ICP-MS. An alpha source was prepared by micro-co-precipitation from the 5 mL 1.5 M HCl strip solution.
Measurements
Liquid scintillation counting (LSC) A Perkin Elmer TriCarb 2800 facility was used to measure the samples of the batch uptake experiments containing single tracer. The k′ capacity factors were calculated against the added activities of the spikes (A0). Standard uncertainties of k′ were 5–10% if k′ was not exceeding 10,000.
Alpha spectrometry (AS) Passivated Ion Implanted Si detectors (AMETEK, type TU-019-300-AS) attached to EG&G Ortec 576A dual alpha spectrometer and EG&G Ortec Ethernim multichannel analyzer were used to collect alpha spectra. Sources were measured in evacuated chambers at an approximate source-detector distance of 5 mm and source diameter of 25 mm. EG&G Ortec Maestro emulation software was used to evaluate the spectra. Typical resolution of the alpha spectra was 40 keV, counting efficiency was 17%. Chemical recoveries relative to the activities of the tracers added were calculated with typical uncertainties of 5–10%.
Atomic absorption spectrometry (AAS) A VARIAN SpectrAA-30 instrument with Fe, Ca and Mg lamps was used for the determination of elemental concentrations of Fe, Ca and Mg, respectively. Concentrations were regarded as information values.
Inductively coupled plasma mass spectrometry (ICP-MS) An Agilent Technologies 8800 Triple Quad ICP-MS–MS was used to follow the behavior of lanthanides on DGA. Concentrations were regarded as information values. Measurements were performed at ISOTOPTECH Rt., Debrecen.
Results and discussion
A novel chromatographic procedure has been developed at our laboratory and published recently [7] for the simultaneous separation of all actinides (U, Th–Np, Pu, Am–Cm) from test solutions using a single DGA resin column. The loading conditions and the elution sequence is described in the Experimental Sect. 2.3. The chromatogram obtained after sequential elution of actinides is shown in Fig. 2. Individual actinide peaks are well separated (with the exception of Np and Th that are co-eluted), and chemical recoveries shown in the figure are high for all actinides.

The method for the determination of the actinide isotopes from radioactive waste samples consisted of sample destruction with mineral acids (a), actinides co-precipitations using ferrous hydroxide (b), oxidation state adjustment of actinides and load preparation (c), chromatographic separation of actinides (d), α source preparation (e). The method gave good results for many waste samples, chemical recoveries were acceptable high (> 50%) and the purity of the individual actinide fractions fit the purposes of α spectrometry.
Simple procedures for actinides in soil and sediment: Procedure I and II
Our aim was to adopt the well tested chromatographic separation for soil and sediment samples. As a desired sample mass 5 g were selected, because it is usually enough to detect the few mBq of artificial actinides (Pu, Am) in a surface soil layer and it is not too lot to detect the typical few hundred mBq of natural actinides (U, Th). For sample destruction we wanted to use fusion because it is much faster and more efficient than acid destruction in the complete dissolution of many silicates, alumina silicates containing natural radionuclides (e.g., uranium, thorium) as mineral inclusions. NaOH fusion in a zircon crucible was selected according to the recommendations of Maxwell et al. [8]. Samples were also destroyed by evaporation with mineral acids (HF, HNO3, HCl occasionally H2O2) in Teflon and glass beakers in order to compare the destruction procedures. To speed up the conventional procedure and to make it more intensive, the destruction was started in a microwave oven followed by evaporation with acids in an open system.
Both destruction techniques were directly combined with the chromatographic separation without any pre-concentrations thus obtaining Procedure I using acid destruction and Procedure II using alkaline fusion (shown in Fig. 3) to obtain rapid and simple methods. Due to the extremely high capacity factors of all actinides on DGA resin from the 4 M HCl load solution in reducing media we expected high recoveries.

Flow-charts of the simple separation procedures for soil or sediment. a Procedure I: Destruction with mineral acids using MW and evaporation and EC chromatography using DGA column, b Procedure II: Destruction by fusion with NaOH and EC chromatography using DGA column
Both procedures seem to be very simple. Complete acid destruction takes almost 3 days while fusion with NaOH is performed during a couple of hours. For preparation of the load samples have to be transferred into 0.5 M HCl. In this media actinides can be reduced using Na2SO3 that proved a good reducing agent for uranium and plutonium and did not interfere with chromatographic separation (not like other reducing agent such as hydrazine).
After acid destruction the residue could be taken up with minimum 50–60 mL 0.5 M HCl. Filtration through 0.45 μm pore size membrane was not fast indicating the presence of some insoluble residue, especially the gel-forming silica. (Most of the silica content of the sample was previously removed by evaporation with 40% HF.) After reduction that took about half an hour the sample was cooled and acidified to 4 M with 37% HCl, thus a load solution of 80–100 mL was obtained that had to be filtered again because increasing the acidity caused additional formation of some silica gel.
After fusion with NaOH the melt was dissolved with limited amount of water (about 100 mL) that meant a NaOH concentration of about 10 M. This solution contained a significant amount of slurry, mainly insoluble hydroxides. The solution was acidified carefully with 37% HCl to about 0.5 M HCl concentration and boiled intensively. A gelatinous solution of about 200 mL was obtained. The silica gel content of the sample had to be removed. Therefore samples were centrifuged producing a significant amount of colorless gel. The supernate was filtered through 0.45 μm pore size membrane, but filtration was slow, in some cases the membrane was completely blocked. To these samples we added polyethyleneglycol (PEG) as coagulant before or after centrifuging. The filtration had to be repeated after oxidation state adjustment and acidification to 4 M. Finally, about 300 mL load solution was obtained, and the whole procedure to remove silica gel took about a day if it did not fail completely.
Both methods were tested with standard reference materials produced by the IAEA, soil samples IAEA-326, IAEA-375 and sediment samples IAEA-300 and IAEA-367. Chemical recoveries obtained in the whole procedure are shown in Table 2.
It can be concluded that acceptable high recoveries (> 50%) were obtained for all four actinides in the soil samples IAEA-375, IAEA-326 in Procedure I, and very similar results were obtained for the same samples in Procedure II. It is amazing that even 300 mL load solution could be passed through the column filled with 0.5 g DGA resin. Theoretically the capacity factors of the 4 actinides on DGA are ≥ 1000 from 4 M HCl what allows their retention from a load solution of ≥ 800 mL. But the samples contain significant amount of various matrix components including Fe that is also well retained from 4 M HCl load. The high actinide retention reveals that at least some matrix elements do not have deleterious effect on separation. The slightly lower recoveries in Procedure II might be attributed to losses of actinides due to their removal together with the lot of silica gel. Recoveries of Pu and Am were also high in case of sediment IAEA-300, but low recoveries were obtained in sediment IAEA-367, and reduction of U and Th recoveries were observed in case of IAEA-300. We were faced with the following issues: (i) Procedure I is slow due to the long destruction and it fails for certain actinides in certain samples, (ii) Procedure II is slow or even blocked due to the lot of silica gel formed in the acidic media and the procedure fails for certain actinides in certain samples. We suspected that there was a matrix dependence in the actinides retention on DGA. Therefore we decided to determine the matrix composition of the four SRMs, and based on that knowledge to develop a modified separation scheme.
Elemental composition of the standard reference materials
Differences in the elemental composition of the samples might give explanation for the differences in recoveries. The major components of soil and sediment are Si, Ca, Al, Fe. Silicon as silica gel is not chemically interfering with the DGA extraction. It is known from the literature [11] that Ca is retained by DGA from nitric acid solutions and it reduces the Am retention, but no Ca retention and no interference of Ca on Am retention in 4 M HCl was proven so far. Calcium has very slight adsorption on DGA from 4 M HCl, but its amount in the samples is big. Alumina is not retained on DGA, but its salts are known as common salting-out agents. Iron is well retained on DGA from 4 M HCl, but from our experiences we know that high amount is tolerated when actinides are retained from 4 M HCl. Other elements that are retained on DGA—according to the basic work of Pourmand and Dauphas [12]—are Zr and lanthanides, but these elements are not typical matrix components in soil and sediment, and their amounts are usually far below the capacity of the DGA column (a few mg referring to o.5 g DGA resin).
Certifying documents of the SRMs were applied to determine the composition of the matrix elements and Zr and Atomic Absorption Spectrometry was used to measure Fe, Ca in some samples in order to establish the data in Table 3.
While SRM soils IAEA-375 and IAEA-326 have similar composition it is clear from Table 3 that IAEA-300 and IAEA-367 are sediments of high Fe and high Ca content, respectively. The basic question is whether these differences in matrix components are responsible for the different retention of actinides on DGA. To give an adequate answer a series of batch uptake experiments were performed with the four actinides from test solutions containing different concentrations of Ca and Fe in 4 M HCl as models of load conditions on DGA. Additionally, the effect of Al and Zr was also studied.
Batch uptake experiments
50–100 mg of DGA resin were equilibrated with 1.5 mL of a 4 M HCl solution spiked with known activities of the radiotracers, i.e. 241Am, 239Pu, 230Th and 233U. Solutions contained various amounts of reagents to be tested. In the first set of experiments, the reducing agent Na2SO3 used for preparation of the load was added to the samples in 0–-62,500 ppm concentration range to see whether the capacity factors of the actinides are changed. The effect of Fe(III) was studied in a similar way adding FeCl3 to the test solutions in 0–4375 ppm Fe concentration. Then the Na2SO3 concentration was fixed at 30,000 ppm, a typical value in load, and the Fe(II) concentration was changed in the 0–4375 ppm range using FeCl3 (that is reduced to Fe(II) during the test). The effect of Ca was studied analogous to Fe, adding CaCl2 to the test solutions in 0–12,500 ppm concentration. Then the Na2SO3 concentration was fixed also at 30,000 ppm, and the Ca concentration was changed in the 0–12,500 ppm range using CaCl2. Finally, Na2SO3 and Fe(II) concentrations were fixed at 30,000 and 2500 ppm, respectively and the Ca concentration was varied in the range of to 0 12,500 ppm. Ranges of each reagent were selected in order to cover the concentration ranges in the load solutions of real soil and sediment samples (compare with Table 3). Capacity factors (k′) calculated for the different test conditions are shown in Fig. 4. Vertical lines indicate the concentrations in the load, where 375 refers to SRM IAEA-375 which is a typical soil sample, 300 and 367 refer to SRMs IAEA-300 and IAEA-367 which are samples of extreme high Fe and Ca content, respectively.

Capacity factors of actinides on DGA resin in presence of possible interferences: a Na2SO3, b FeCl3, c Na2SO3 + FeCl3, d CaCl2, e Na2SO3 + CaCl2, f Na2SO3 + FeCl3 + CaCl2. Dotted lines represent values below detection limit (LD)
The effect of the reagents on actinide retention was classified in three groups: reagents of no effect, reagents producing synergistic effect by increasing k‘and those of antagonistic effect reducing the k‘values of actinides. From Fig. 3a it is clear that the presence of Na2SO3 has a synergistic effect on all actinide retention in a wide concentration range. It is especially effective for trivalent actinides (Am, Pu(III)). The role of Na2SO3 is reduction and increasing the k′ values. Fe(III) improves the retention of Am and Pu (here Pu is likely to be in various oxidation states), but it reduces k′ in case of Th and especially U (that is probably hexavalent) (see Fig. 3b). When Na2SO3 and Fe are present together than actinides and Fe(II) are in reduced forms, but the synergistic (Am, Pu(III)) and antagonistic (Th, U(IV)) effects are very similar than in case of Fe(III) (see Fig. 3c) This figure allows us the conclusion that in presence of high Fe concentration like in IAEA-300, uranium is not retained on the DGA column, while in IAEA-375 of smaller Fe content uranium retention is still sufficient.
Calcium reduces the retention of all actinides but it has antagonistic effect especially for Am and Pu (that is a mixture of various oxidation states) (Fig. 3d). When Na2SO3 and Ca are present together, actinides are in reduced forms, and k′ values are also reduced (Fig. 3e). If Ca concentration is below 1000 ppm in the load, what is the case for typical soil samples like IAEA-375 than k′ is still big enough for complete retention. At higher Ca concentrations the k′ values for Am and Pu(III) become much smaller probably causing that they are not retained on DGA. Thorium and U can be retained even from samples of extreme high Ca content such as IAEA-367.
In summary, it can be concluded that extreme high Fe(II) and Ca concentrations are antagonistic for the retention of U, Th and Am, Pu, respectively. The last Fig. 3e shows that in case of high Fe, Ca and Na2SO3 concentrations U and Th cannot be retained while the retention of Am and Pu is still possible if load volume is limited to our standard conditions of 100–200 mL.
The batch uptake experiments of actinides on DGA in presence of AlCl3 (0–6250 ppm Al) and ZrOCl2 (0–5000 ppb Zr) showed that k′ values were not affected.
In order to get high actinide retention from about 100 mL load solution it is desirable that the k′ capacity factors should exceed 100 and the load solution should have the following concentration of the matrix components:
- Na2SO3:
-
about 30,000 ppm to assure the strong reducing conditions even if a part of Na2SO3 is decomposed in the 4 M HCl solution;
- Fe2+:
-
between 100 and 1000 ppm, more than 100 ppm helps adjust reducing conditions and less than 1000 ppm avoid the reduction of k′ factors for U and Th (addition of 0.5 g of Mohr’s salt to the load solution will meet this requirement if it has originally neglectable Fe content);
- Ca2+:
-
less than 2000 ppm to avoid the reduction of k′ factors for Pu and Am, it is desirable to minimize the Ca content in the load;
- Al3+:
-
does not have an impact on actinides retention on DGA;
- Zr4+:
-
does not have an impact on actinides retention on DGA
It is very important to note that Fe and Ca strongly affect the retention of actinides on DGA, and the effect is different on the trivalent (Am, Pu(III)) and the tetravalent (Th, U(IV)) actinides. Iron that is retained itself on the resin, improves significantly the retention of the trivalent actinides probably forming a special Ac(III)-Fe(II)-chloro-DGA complex. The situation is completely different for the tetravalent actinides probably forming Ac(IV)-chloro-DGA complexes that may be displaced by the Fe-chloro-DGA complexes. The situation with Ca is also difficult to understand because Ca-chloro-complexes are very slightly or not at all retained by the DGA (k′ < 10) from 4 M HCl, nonetheless, they displace all actinides especially the trivalent ones if the Ca concentration is high. We can imagine that binding sites for tri- and tetravalent actinides in DGA are different. Unfortunately, we do not have tools to test the mechanism of actinide binding to DGA.
Development of procedure III
According to the results of the batch uptake experiments a new procedure was planned where sample pretreatment (actinide pre-concentration) steps were included between fusion and chromatographic separation. Our aim was to adjust the Fe concentration in the optimized region, minimize the Ca concentration and remove the silica content. We did not want to use additional chromatographic separation in the pre-treatment to keep the procedure simple and cheap, instead we wanted to fulfill our goals using a combination of co-precipitations. Our new concept was the following:
-
To remove silica there are two simple techniques. In the concentrated NaOH solution that is obtained after dissolution of the fusion cake with water, silica forms soluble sodium silicate that can be easily removed by fast filtration. Actinides usually remain in the mixed hydroxide precipitate. Addition of some Fe(II) and hydrazine as reducing agent will help retain also the penta- or hexavalent actinides such as uranium in the precipitate. Silica remaining in the precipitate will form gelatinous silica gel if the sample is acidified, but silica residues can be removed by the addition of concentrated HF when soluble H2SiF6 is formed. HF will form insoluble alkaline earth fluorides that can carry actinides under reducing conditions. Addition of some Fe(II) and hydrazine will assure this condition. Filtration that is typically fast will separate the precipitate carrying the actinides from the rest of silica as fluoride. The fluoride precipitate can be dissolved with boric acid in the presence of nitric or hydrochloric acid because fluorides can form soluble [BF4]- complex.
-
To reduce (optimize) the Fe concentration of the sample, the precipitation of fluorides is an excellent tool because Fe forms soluble fluoride complexes such as [FeF6]3− and [FeF6]4−. While filtering the alkaline earths fluorides with retained actinides most of the iron is automatically removed.
-
Calcium removal seemed to be the most problematic. For reduction of the Ca concentration we considered 3 options.
-
(a)
Making an additional ferrous hydroxide co-precipitation of actinides from a big volume (2–3 L) of boric acid containing solution, because the solubility of Ca(OH)2 is higher than that of Fe(OH)2 at about pH 7. Thermodynamic calculations showed that the Ca concentration can be reduced by a limited amount and a big volume of solution had to be filtered that took longer time.
-
(b)
Making an additional sub-stoichiometric CaF2 co-precipitation. If actinides are carried with the small amount of CaF2 precipitate than part of Ca remaining in solution is separated by filtration. Model experiments were planned to check the retention of actinides with the small amount of fluoride precipitate. (Later it was proven that uranium and americium were not completely carried with the precipitate resulting in low recoveries for U and Am.)
-
(c)
Making an additional sub-stoichiometric Ca(OH)2 co-precipitation in the presence of Mg(OH)2. From the experiment described above (in point b) it turned out that only bigger amount of precipitate can carry all actinides. Therefore we selected Mg(OH)2 as an appropriate precipitate to carry all actinides. Contrary to Ca it is likely that Mg will not interfere with the separation of actinides on DGA, because it is not at all retained by DGA. The solubility product of Mg(OH)2 is much lower (Ksp = 5.61E − 12) than that of Ca(OH)2 (Ksp = 5.5E − 6), thus Mg will precipitate before Ca starts to precipitate as hydroxide. With calculated amount of sodium hydroxide Mg(OH)2 can be precipitated while Ca remaining in solution is removed by filtration. To assure actinide co-precipitation with Mg(OH)2 some Fe(II) is added to the solution to assure reducing conditions. Model experiments are to be performed to check (1) the effect of Mg on the actinide separation on DGA, (2) the retention of actinides on Mg(OH)2 precipitate, (3) the concentration of Ca in the load after Mg(OH)2/Ca(OH)2 co-precipitation.
-
(a)
The flowchart of the latter procedure is shown in Fig. 5.

Flowchart of procedure III
Calcium removal by co-precipitation of actinides with sub-stoichiometric CaF2 precipitate
We tried to co-precipitate actinides with sub-stoichiometric amounts of CaF2 precipitate to reduce the Ca content of the samples.
To simulate the conditions of treating 5 g of sample of high Ca content we assumed that samples have a maximum of 1.5 g Ca content that is equal to 0.075 mol Ca per sample. It was assumed that after silica gel removal the residue was dissolved in 100 mL boric acid containing solution. Tests were performed with this simulated stock solution after 10 times volume reduction. The Ca stock solution was prepared as described in CaF2 test in Experimental Sect. 2.2. 10 mL aliquots were spiked with 239Pu, 241Am, 233U, 230Th. 40% HF was added to the test solutions in various amounts to simulate sub-stoichiometric precipitation of CaF2. Samples were equilibrated, then centrifuged and aliquots of the supernates were analyzed by LSC. Results of analysis as % of activity retained by the precipitate were calculated. The amount of Ca in the supernate was determined by AAS of blank test (not spiked Ca stock solution) and the % of Ca precipitated was also calculated. Results of LSC and AAS measurements are summarized in Table 4.
We found that recoveries of Am and U(IV) were low (≤ 21%) when the amount of CaF2 precipitate was reduced to less than 9 and 29% of the original Ca amount, while those of Th and Pu(IV) were acceptable high if Ca content in the precipitate was > 29%. Sub-stoichiometric CaF2 precipitation is not adequate for concentration of all actinides.
Calcium removal by co-precipitation of actinides with sub-stoichiometric Ca(OH)2 precipitate in presence of excess Mg(OH)2
-
(1)
The effect of Mg on the actinide separation on DGA
Before testing actinides co-precipitation with Mg(OH)2 we checked the effect of Mg on the separation of actinides on DGA. We performed 2 chromatographic separations with 70 mL load solutions spiked with a mixture of actinides (239Pu, 241Am, 233U, 230Th). Load solutions contained equivalent amounts of Ca or Mg (0.0375 M). The separation was performed according to the standard protocol described in Experimental Sect. 2.3. Chemical recoveries are given in Table 5.
Table 5 Chemical recoveries of actinides after separation on DGA column Results show that the recoveries of Pu and Am are low if load solution contains high concentrations of Ca, how it is expected from the batch experiments, too, while the presence of Mg does not affect the separation.
-
(2)
The retention of actinides on Mg(OH)2 precipitate
We tried to co-precipitate actinides with Mg(OH)2 while the precipitation of Ca as hydroxide is limited (sub-stoichiometric). Therefore we performed tests with model solutions where Mg was added to the sample in bigger amount.
A Mg stock solution of the same molarity as that of the Ca stock solution was used (see Mg(OH)2 test in Experimental Sect. 2.2). To the 10 mL solution which contained boric and nitric acids and individual actinide spikes (239Pu, 241Am, 233U, 230Th) 5 M NaOH was added in various amounts to simulate sub-stoichiometric precipitation of Mg(OH)2. Samples were equilibrated, then centrifuged and aliquots of the supernates were analyzed by LSC. Results of analysis as % of activity retained by the precipitate were calculated and summarized in Table 6.
Table 6 Retention of actinides on different amounts of Mg(OH)2 precipitate Addition of only 500 μL of 5 M NaOH results in high recoveries for all actinides. This NaOH amount is about the one-third of total amount of NaOH needed to precipitate all Mg as hydroxide, and the pH of the test solution is still acidic. It is likely that only a small amount of Ca will co-precipitate with Mg under such conditions. To determine the actual amounts further tests were performed.
-
(3)
The concentrations of Ca and Mg in the mixed Mg(OH)2/Ca(OH)2 precipitate
Two series of tests were performed using the Ca stock solution. In the first one (only Ca present), the 10 mL aliquots were used as they were received, in the second one (Ca and Mg present) 84 mg Mg as chloride salt were added to each sample. The molarities of Ca and Mg were equal. Then the NaOH solution was added to the samples and the tests were performed as described in the Experimental Sect. 2.2 (see Ca(OH)2 test and mixed Mg(OH)2/Ca(OH)2 test). Results of the AAS measurements are shown in Table 7.
Table 7 Ca and Mg content of the precipitates in the Ca(OH)2 test and the mixed Mg(OH)2/Ca(OH)2 test
It is clearly seen from the table that the amount of Ca precipitated as hydroxide is significantly reduced (using 1500 μL 5 M NaOH from 65% to 14% of its original amount) if Mg is added in excess to the solution. The amount of Mg in the precipitate is about 67% of the total Mg content. This amount of Mg(OH)2 precipitate will carry all actinides as it was shown in the previous test (2). Therefore we decided to use the procedure shown in Fig. 5 for the determination of actinides in soil and sediment samples.
Determination of actinides in standard reference materials by procedure III
Procedure III that consists of sample destruction by fusion (a), actinides co-precipitations using ferrous hydroxide (b), mixed fluorides (c) and mixed (magnesium) hydroxides (d), oxidation state adjustment of actinides and load preparation (e), chromatographic separation of actinides (f), α source preparation (g) was tested using the SRM samples of the IAEA: IAEA-375, IAEA-326, IAEA-300 and IAEA-367. Procedure III is described in detail in the Experimental Sect. 2.4.3. Results are shown in Table 8.
Chemical recoveries of U, Pu and Am for the combined procedure were high, the average values were 76, 74 and 96%, respectively. High recoveries were obtained for the sediments of high Fe and high Ca content, showing that the pre-concentration steps were efficient. Thorium recoveries are surprisingly low, the method is not adequate for Th determination. We suspect that hydrolysis and/or adsorption of Th species are responsible for the losses.
The procedure is relatively fast and robust, it can be performed during 2 days. Silica removal has been performed successfully, the precipitates are usually easy to filter.
Separation of lanthanides from americium
Soil and sediment samples contain lanthanum and lanthanides typically more than 100 μg per gram of sample. Lanthanides (Ln) are retained by the DGA together with americium. If they are stripped also together then the α source prepared for Am analysis from 1 g of sample is already too thick to get good resolution. Therefore an Am lanthanide separation is needed that is usually performed by chromatography on anion exchanger or TEVA resin [14].
DGA resin has a relatively high selectivity for americium against heavy lanthanides and a moderate selectivity against a number of light lanthanides how it is shown by the capacity factors determined by Horwitz et al. [6] and Pourmand et al. [12]. A procedure for the chromatographic separation of lanthanides from each other on a long DGA column (length of 28 cm) has been developed by Párkányi et al. [13]. Based on this procedure we adopted an elution sequence for our standard DGA column (length of about 30 mm). An elution test was performed with a model solution of 15 mL 4 M HCl that contained 0.1 μg of the following elements: La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu (totaling 1.4 μg). The standard eluent for stripping Am that co-elutes all lanthanides is 15 mL 0.5 M HCl (after 5 mL 0.5 M HNO3 wash solution). These solutions were replaced by the set of the following eluents: 10 mL 0.5 M HNO3, 3 mL 0.05 M HNO3, 5 mL 1.5 M HCl. Each mL of the eluates was analyzed for lanthanides by ICP-MS. The chromatograms are shown in Fig. 6.

Elution chromatograms of lanthanides from standard DGA column as a function of the composition and volumes of the eluates
Since Am behaves very much like Nd or Sm we regarded the eluent of 5 mL 1.5 M HCl as the Am fraction. To determine the lanthanide content of the Am fraction of a real sample, we performed the lanthanide analysis of the IAEA-300 sediment. 5 g of sample were processed as described in Experimental Sect. 2.4.3. 100 mL load solution was passed through the standard DGA column, and the chromatography was performed as usual with the modified elution sequence: 10 mL 0.5 M HNO3, 3 mL 0.05 M HNO3, and finally stripping Am with 5 mL 1.5 M HCl. Lanthanides in the eluent were measured by ICP-MS. Chemical recovery of Am in the Am strip solution was calculated from measurement of the alpha source prepared by micro-co-precipitation (Table 9).
In the Am source the total amount of lanthanides was reduced to about one-third of the original value. In presence of about 100 μg of lanthanides Am sources of good resolution can be prepared, and the peaks of 241Am and 243Am are well separated. Further chemical processing is not required. This was the situation in case of the 4 tested reference materials. Americium recoveries were always high (≥ 92%).
Conclusions
A fast, robust and cheap combined method has been developed for the simultaneous determination of uranium, plutonium and americium nuclides in soil and sediment. Samples up to 5 g are destroyed by fusion with sodium hydroxide. Actinides are pre-concentrated by three consecutive co-precipitations (mixed hydroxides, fluorides, hydroxides) where silica is removed as soluble sodium silicate and silicon fluoride complex, iron is removed as soluble iron fluoride and calcium concentration is reduced by sub-stoichiometric precipitation of calcium hydroxide in presence of magnesium hydroxide. According to a series of tests it was proven that actinide recovery in the pre-concentration is high (> 75%). By applying the special sample preparation procedure the chromatographic separation of actinides on DGA became almost matrix independent.
The separation of actinides is performed on a single small DGA resin column by sequential elution where the separation of americium from light and heavy lanthanides (La, Ce, Pr, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu) also takes place. Alpha sources prepared from the individual actinides strip solutions have good resolution, no contamination is detected by alpha spectrometry. Chemical recoveries of U, Pu and Am for the whole procedure are high (the average is higher than 75%). The whole procedure can be determined in 2 days. The procedure failed for the separation of thorium.
The method can be used for determination of alpha emitting uranium, plutonium and americium nuclides in environmental samples (above 0.1 Bq/kg) for routine monitoring purpose, as well as in contaminated samples of elevated activities for emergency response or decommissioning purposes.
References
Lehto J, Hou X (2011) Chemistry and analysis of radionuclides. Wiley-VCH Verlag, Weinheim
EiChrom Technologies Inc homepage. https://www.eichrom.com/eichrom/products/
TRISKEM International homepage. https://www.triskem-international.com/ resins-and-accessories.php
Vajda N, Torvenyi A, Kis-Benedek G, Kim CK, Bene B, Macsik Z (2009) Rapid method for the determination of actinides in soil and sediment samples by alpha spectrometry. Radiochim Acta 97(8):395–401
Bochicchio F, Chalupnik S, Hampe D, Kim CK, Kis-Benedek G, Kleinschmint R, Törvényi A, Sansone U, Vajda N (2009) A procedure for the rapid determinatiion of Pu isotopes and Am-241 in soil and sediment samples by alpha spectrometry. IAEA Anal Qual Nucl Appl Ser IAEA/AQ 11:1–39
Horwitz EP, McAlister DR, Bond AH, Barrans Jr RE (2005) Novel extraction of chromatographic resins based on tetraalkyldiglycolamides: characterization and potential applications. Solvent Extr Ion Exchange 23:319–344
Groska J, Vajda N, Zs Molnar, Bokori E, Szeredy P, Zagyvai M (2016) Determination of actinides in radioactive waste after separation on a single DGA resin column. J Radioan Nucl Chem 309(3):1145–1158
Maxwell SL (2008) Rapid method for determination of Pu, Am and Cm in large soil samples. J Radioan Nucl Chem 275(2):395–402
Luisier F, Alvarado AC, Steinmann P, Krachler M, Froidermax P (2009) A new method for the determination of plutonium and americium using high pressure microwave digestion and alpha spectrometry or ICPMS. J Radioan Nucl Ch 281:425–432
Eikenberg J, Jaggi M, Beer H, Röthi M, Zumsteg I (2009) Separation techniques for low-level determination of actinides in soil samples. Appl Radiation Isotopes 67(5):776–780
Daum JK, Sudowe R (2018) Calcium interference on americium and plutonium uptake on six extraction chromatographic resins. J Radioan Nucl Ch 318:331–339
Pourmand A, Dauphas N (2010) Distribution coefficients of 60 elements on TODGA resin: application to Ca, Lu, Hf, U and Th isotope geochemistry. Talanta 81(3):741–753
Párkányi D, Szentmiklosi L, Vajda N (2017) Lanthanide and americium separation with DGA resin. In: 26th Seminar on activation analysis and gamma spectrometry (SAAGAS 26)
Vajda N, Kim CK (2010) Determination of Am-241 isotope: a review of analytical methodology. J Radioan Nucl Chem 284(2):341–366
Acknowledgements
The authors would like to thank Prof. Steffen Happel, TRISKEM International for valuable advices, Mihály Veres, PhD, ISOTOPTECH Rt. for ICP-QQQ-MS measurement possibilities and Patricia Szeredy for participating in the chemical work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Vajda, N., Zagyvai, M., Groska, J. et al. Determination of uranium, plutonium and americium in soil and sediment by a sequential separation procedure using a single DGA column. J Radioanal Nucl Chem 326, 695–710 (2020). https://doi.org/10.1007/s10967-020-07337-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-020-07337-9