
Abstract
The presented study reports on the purification of molybdenum oxide, which is one of the important tasks of the Advanced Mo based Rare process Experiment in searching for the neutrinoless double beta (0νββ) decay of 100Mo. Purified MoO3 powder is used as initial material for further growth of radiopure monocrystals. As purification technique, double sublimation, co-precipitation with calcium chloride carrier, and precipitation of polyammonium molybdate from acidic media were used. Concentrations of impurities like Sr, Ba, Pb, Th and U were measured by ICP-MS and radioactive isotopes were checked by a HPGe detector at the YangYang underground Laboratory in Korea.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The neutrino oscillation experiment reveals the existence of non-zero neutrino mass [1]. After these results, the next things to find are the mass value of neutrino and its nature, if it is Dirac or Majorana particle [2]. At present time, different collaborating groups are searching for the 0νββ decay using the different isotopes 76Ge, 130Te, 136Xe, 82Se, 150Nd, 100Mo, 48Ca, and 116Cd [3,4,5,6,7]. Among those groups, the AMoRE (the Advanced Mo based Rare process Experiment) collaboration [8] is searching for the 0νββ decay of 100Mo using radiopure scintillating crystals based, mainly, on the 40Ca100MoO4 and also on Li 1002 MoO4, Na 1002 Mo2O7 single crystals. Since, 100Mo is one of the most promising nuclei, owing to its high transition energy (Q ββ = 3034 keV), comparative ease of enrichment by centrifugation, which provides extremely high purity level of the obtained product [9, 10].
The AMoRE project is a series of experiments. The first phase of the AMoRE, called AMoRE-Pilot, has been operating since 2015 with about 1.5 kg of 40Ca100MoO4 crystals at YangYang underground Laboratory (Y2L) in Korea. The second phase of the experiment, AMoRE-I with 4.5 kg of 40Ca100MoO4 crystals, is planning to begin in 2018, and a half-life sensitivity of 2.7 × 1025 years, which corresponds to the effective Majorana neutrino mass to be 70–140 meV, is expected. The third phase of the experiment called AMoRE-II will begin operation in 2020 with about 200 kg of Mo-based crystals with the decision on the type of crystal still to be made. With operating the experiment for 3 years, the expected half-life is 1.1 × 1027 years corresponding to the effective mass sensitivity of 12–22 meV. The background levels for the AMoRE-I and -II are required to be 0.001 and 0.0001 counts/keV kg year in the region of interest, respectively.
The sensitivity of the detector used by the AMoRE is determined by the background in the region of the expected peak [11]. The major sources for such background are due to decay chains of natural isotopes, mainly Ra, Th and U, which are present as impurities in detector material [12, 13]. These isotopes have higher decay energy than 3034 keV. The reduction of these isotopes assures reaching of the required internal background level of molybdenum based scintillating bolometers.
As was mentioned above, enriched 100Mo is manufactured by the centrifugal method. As a result of isotopic enrichment by centrifugation, purification from heavier than 100MoF6 gas fractions, such as WF6, UF6 and ThF6, occurs, that’s why obtained after hydrolysis and calcination 100MoO3 powder (99.997%) is already pure in comparison to the natural one and could be used for first phase of the experiment [14]. While commercially available natural molybdenum trioxide is used for the investigation of growing of molybdenum-based monocrystals, for obtaining of preliminary scintillation properties of the crystals at room and cryogenic temperatures. In order to get adequate results, this commercial MoO3 powder should have the same purity level as the enriched one. Moreover, investigation of techniques for purification of raw MoO3 would help for further investigation of deep purification even of enriched one.
With above qualification in mind, there are stringent requirements applicable to the scintillators: the absence of paramagnetic elements, high light yield and low levels of internal radioactive background. After purification of initial components and further crystal growth, the final background level of the scintillating bolometer should be at most 10−5 Bq/kg, and content of other impurities below 0.1 ppm is also important to make a high light yield crystal. Preliminary experience for growing of monocrystals has shown that crystal growth gives about a factor of 10 reduction for those backgrounds, so our goal for backgrounds are about 1 ppt for Th and U and μBq/kg for Ra in Mo-based monocrystal. Therefore, the background levels should be at least 10 ppt for Th and U and 10 μBq/kg for Ra. Thus, in order to achieve the established requirements, it is critical to improve the known methods and develop new approaches for deep purification of initial materials.
Experimental
Equipment and reagents
As initial material, commercial molybdenum tri-oxide powder 99.95% purity grade produced by Alfa-Aesar was used. Similarly, the NH4OH solution (~ 25% Puriss), produced by Sigma Aldrich was used as a main solvent. High purity deionized water (resistivity of 18.2 M Ω cm−1) obtained from Milli-Q water purification was used for preparing samples and solutions. Hydrochloric acid (30%) of High Pure Analytical Reagent produced by Eco Research Inc. was used. As a co-precipitating reagent 10% calcium chloride solution was prepared by dissolving of commercial carbonate CaCO3 powder of 99.997% purity grade (Alfa-Aesar) in a small volume of hydrochloric acid and diluting with water.
All the tests were carried out inside a clean room (class 1000). For doing the experiments a high quality quartz beaker and crucible were used. Before every test the apparatuses were washed with 1% HNO3 using sonication and then with di-water.
The inductively coupled plasma mass spectrometer (ICP-MS) equipped with a collision cell (Agilent 7900, Agilent Technologies, USA) operating with high-purity helium was used for the detection of Sr, Ba, Pb, Th and U concentrations. The method detection limit for tracers of U and Th refers to 20.0 ppt in MoO3 powder. Prior measurements the molybdenum samples were dissolved up to 1% MoO3 by mixture of ammonium hydroxide (0.3%) and hydrochloric acid (1.0%), the final molybdenum concentration of no more 0.066 g Mo/mL. Operating in helium collision mode and ultra high matrix introduction (UHMI) was used for reduction of polyatomic interferences and matrix effect.
Measurements of radioactivities were done by a high purity germanium detector (HPGe) placed at the Y2L in South Korea. Molybdenum samples were placed in containers with a Marinelly geometry to optimize the detection efficiency. The detection limit for HPGe detector is about 1.0 mBq/kg with a 1 kg sample for measurement of 1-month period.
Sublimation procedure
For the experiment, initial MoO3 powder was kept inside the quartz tube and sublimated at the temperature 720 °C under vacuum (< 0.013 mbar). Temperature of the desublimation was kept around 550–575 °C.
Preparing of the initial molybdenum solution
In order to prepare the initial ammonium molybdate (AM) solution, the raw MoO3 powder was completely dissolved in aqueous ammonia solution and filtered using a 0.1 µm size pore membrane filter with a view to separate insoluble impurities.
Wet chemistry technique. Co-precipitation with CaCl2
Required amount of 10% CaCl2 solution was added into initial Ammonium Molybdate (AM) solution in such a way as to precipitate out 3% of molybdenum in the form of calcium molybdate. Precipitate was separated using membrane filtering. The polyammonium molybdate (PAM) powder as a final product was obtained by precipitation with hydrochloric acid within acidic media at pH 1.8 and washing with 10% pure NH4Cl solution.
Results and discussion
Effectiveness of purification by law vacuum sublimation
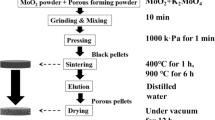
Sublimation of MoO3 under atmospheric pressure is widely used as a purification technique and based on phase transition of a substance from solid to gas phase avoiding melting [15]. Higher temperature of sublimation requires less time for the complete process. But overheating may cause melting of molybdenum oxide and hence low yield or effectiveness of purification [16, 17]. In order to ascertain the optimum temperature of sublimation for the highest effectiveness of purification and yield of purified product, the sublimation studies were performed for 50 g of raw molybdenum oxide for the temperature range of 670–750 °C.
The results shown in Fig. 1 indicate that single sublimation is very effective purification technique for a lab scale application. Moreover, purification by sublimation provides high yield efficiency above 94%. At present experiment, the main parameter for the determination of optimal temperature was barium reduction, so in Fig. 1 calculated decontamination factor (DF) are presented. The DF value was calculated as ratio of Ba concentration in initial raw MoO3 to Ba concentration in the purified powder.

Barium decontamination factors obtained at different sublimation temperatures
Thus, the highest values of decontamination factor (114) and the yield of purified product (97%) were obtained at the temperature of 720 °C. Taking into account the time required for sublimation, 720 °C was chosen for the next studies.
For the further experiment, 1.2 kg of initial MoO3 powder was sublimated successfully twice at temperature of 720 °C. Obtained decontamination factors and impurities levels are presented in Table 1.
As one can see from the data presented in Table 1, using of successive double-sublimation of molybdenum oxide allows to improve purification by one to two orders of magnitude and to achieve decontamination factor of ~ 80 for Sr and Ba, at least larger than 7 for Th and ~ 60 for U. Due to low sublimation temperature of lead already at 400 °C, decontamination from Pb could not be achieved properly.
Since the concentration of radium (226Ra) and other radioactive elements could not be measured by ICP-MS, the radioactivity level of the obtained double sublimated molybdenum oxide has been measured by a HPGe detector located at the Y2L in Korea (Table 2).
From results shown in Table 2 it is immediately obvious that low vacuum sublimation is effective technique for removing Ra, Ac and K. It is important to observe that the specific activity of 226Ra (from U chain) was reduced 13 times after single sublimation and 80 times after double sublimation, that is almost corresponding to Sr and Ba reduction factor. Since Sr, Ba and Ra belong to the same group of the Periodic Table and have similar chemical behaviour, the decontamination factor for either Sr or Ba can be used as one for 226Ra. Also, the specific activity of 228Ac (from Th chain) after single and double sublimation was reduced 17 and 127 times, respectively. In case of 40K, the reduction factor is 10, and already after single sublimation the specific activity level could not be decreased below 73 mBq/kg.
Effectiveness of purification by law vacuum sublimation
Co-precipitation method with small amount of carrier and precipitation are traditional widely used techniques in molybdenum metallurgy for purification of raw material or ores [18, 19].
Co-precipitation for the purpose of separation of impurities from ammonium molybdate solution with various carriers has been reported [11, 13, 20, 21]. At present study to co-precipitate alkaline earth elements and actinides, calcium molybdate was used as the collector (carrier). Concentrated hydrochloric acid and aqueous ammonia solution were used for the pH-adjustment.
While dissolving of MoO3 in excess amount of ammonia, the normal molybdates occurs:
During dissolving in ammonia at the pH range of 9–10, the impurities existing in initial molybdenum trioxide powder forms insoluble thorium and uranium hydroxides which are partially separated by vacuum filtration with 0.1 µm size pore PTFE membrane filter. On the other hand, ammonium molybdate solution could dissolve low concentration of oxides and hydroxides of many elements, including strontium, barium and lead.
To separate soluble impurities from ammonium molybdate solution, calcium chloride was used as the co-precipitation agent. At basic pH calcium chloride reacts with ammonium molybdate forming calcium molybdate precipitate:
That calcium molybdate precipitate as a collector initiates co-precipitation of Sr, Ba and Ra molybdates. Therefore, calcium chloride solution (10%) was dropwise added into initial filtrated AM solution at pH 7.0 in such a way as to precipitate 3% of molybdenum, and solution was stirred for 6 h. After few hours of exposition CaMoO4 precipitated occurs and sorbs impurities in molybdate form. Then ammonia was added to the solution in order to increase pH up to 9.5–10.0, that strong basic pH provides precipitation of the remaining U and Th and simultaneous sedimentation with carrier. Finally, solution was filtered out through the membrane filter.
In order to separate molybdenum matrix from ammonia solution and the remaining Ca, precipitation of polyammonium molybdate from acidic media by adding HCl is more preferable way in comparison to complete evaporation of solvent or fractional recrystallization. The point is that crystallization of new recrystallized PAM crystals carried out from basic media, as a result, co-crystallization of left amount of impurities and Ca could occur.
Finally, the concentrated hydrochloric acid was added drop by drop with stirring to the obtained after co-precipitation ammonium molybdate solution up to pH 1.8 at the temperature of 40–50 °C to precipitate polyammonium molybdate (PAM) sediment.
After filtration, the PAM powder was washed with pure 10% NH4Cl solution in order to reduce strong acidic pH level and remaining of impurities. The results obtained after purification by precipitation techniques are shown in Table 3.
To estimate effectiveness of purification, decontamination factors and recoveries of diverse elements were calculated. The recovery was calculated as a ratio of the impurities co-precipitated with collector to the initial total amount of impurities:
In such a way, after co-precipitation with calcium molybdate collector initial barium, strontium and uranium contamination was reduced above 96%. Comparably low level of strontium recovery could be coursed by presence of ammonia chloride, which occurs as a result of reaction of hydrochloric acid and ammonia during pH adjusting. In case of thorium, after chemical purification the concentration was decreased below detection limit of ICP-MS measurement (Table 4).
Based on the evidence results, total decontamination factors after applying of combined co-precipitation and precipitation of PAM from acidic media for Sr, Ba, Pb and U were reached up to level of hundreds. Strontium, barium and radium belong to same group of Periodic Table of the Elements and have same chemical behavior and distribution during precipitation. So, knowing effectiveness of purification for Sr and Ba, we could estimate how reduced radium contamination.
In Table 5, for comparison, contamination levels of initial molybdenum oxide, enriched 100MoO3 (99.997%) and molybdenum samples after different purification techniques are shown.
As Table 5 shows, contamination level of the obtained molybdenum samples after combined precipitation technique is sufficiently closed to level of enriched molybdenum oxide, for Pb and U concentration in chemical purified samples are even lower than enriched one. Content of U and Th in the purified molybdenum sample refers to level below 30 ppt, which could be proved in final single crystal by next crystal growth.
Conclusions
Different purification techniques have been used in order to reduce the initial contamination of natural molybdenum trioxide powder, such as low vacuum sublimation and co-precipitation combined with precipitation of polyammonium molybdate from acidic media. Successful double sublimation provided reduction of radioactive contamination on the level above 80 for 226Ra and 120 times for 228Ac, concentrations of Ba and Sr were reduced 80 times as well. The yield efficiency after sublimation of 1.2 kg of MoO3 powder was larger than 99%. Co-precipitation with calcium molybdate collector allows to remove more than 95% of Ba, Pb and U contaminations. The combination of co-precipitation and precipitation allowed to achieve total decontamination factor for Sr, Ba, Pb and U on the level of several hundreds, concentration of thorium was reduced below detection limit of the ICP-MS measurement. Taking into account 3% losses of Mo with collector for co-precipitation, the final yield efficiency after wet chemistry purification technique is around 95%.
Thus, molybdenum samples obtained after chemical purification technique, including co-precipitation with calcium chloride, is sufficiently closed to enriched 100MoO3 and could be used for the next experiments for the searching for neutrinoless double beta decay. Due to detection limits for Th and U with ICP-MS measurement after the wet chemical purification as shown in Table 3, we have not fully demonstrated the requirement of radio-impurity levels for the AMoRE-II but combination of double sublimation with wet chemical purification looks promising and will be applied in order to improve the presented results. Our research for advanced purification technique as well as for lowering the detection limits for ICP-MS measurement is ongoing in parallel.
References
Fukuda S et al (2001) Solar 8B and Hep neutrino measurements from 1258 days of super-kamiokande data. Phys Rev Lett 86(25):5651–5655
Mohapatra RN (2007) Theory of neutrinos: a white paper. Rep Prog Phys. doi:10.1088/0034-4885/70/11/R02
Giuliani A, Poves A (2012) Neutrinoless double-beta decay. Adv High Energy Phys. doi:10.1155/2012/857016
Gando A et al (2016) Search for Majorana neutrinos near the inverted mass hierarchy region with KamLAND-Zen. Phys Rev Lett. doi:10.1103/PhysRevLett.117.082503
Abgrall N et al (2014) The Majorana demonstrator neutrinoless double-beta decay experiment. Adv High Energy Phys. doi:10.1155/2014/365432
Alduino C et al (2017) CUORE sensitivity to 0νββ decay. Eur Phys J C. doi:10.1140/epjc/s10052-017-5098-9
Arnold R et al (2015) Result of the search for neutrinoless double-β decay in 100Mo with the NEMO-3 experiment. Phys Rev D. doi:10.1103/PhysRevD.92.072011
Alenkov V et al (2015) Technical design report for the AMoRE 0νββ decay search experiment. https://arxiv.org/pdf/1512.05957.pdf. Accessed 5 Sep 2017
Rahaman S (2008) Q values of the 76Ge and 100Mo double-beta decays. Phys Lett B. doi:10.1016/j.physletb.2008.02.047
Meija J et al (2013) Isotopic compositions of the elements 2013 (IUPAC technical report). Pure Appl Chem. doi:10.1515/pac-2015-0503
Alenkov VV (2013) Ultrapurification of isotopically enriched materials for 40Ca100MoO4 crystal growth. Inorg Mater. doi:10.1134/S0020168513120029
Firestone RB (1996) Table of isotopes, 8th edn. Wiley, New York
Shlegel VN, Berge L, Boiko RS (2014) Purification of molybdenum oxide, growth and characterization of medium size zinc molybdate crystals for the LUMINEU program. EPJ Web Conf. doi:10.1051/epjconf/20136503001
Shubin A, Kulinich Yu, Skorynin G et al (2006) Gas centrifuges in the production of high-purity volatile substances. In: Proc. XI Int Sci Conf physicochemical process in the selection of atoms and molecules and in laser, plasma, and nanotechologies, TsNIIATOMINFORM, Zvenigorod (Rus)
Somorjai G-A (1968) Mechanism of Sublimation. Science 162:755–760
Clarence D, Chiola V (1968) Process for purifying molybdenum trioxide. Patent no. US3393971 A. Patented 23 Jul 1968
Lu WA, Zhang GH, Jie DA, Chou KC (2015) Oxidation roasting of molybdenite concentrate. Trans Nonferr Met Soc. doi:10.1016/S1003-6326(15)64067-5
Kirby HW, Salutsky Murrell L (1964) NAS-NS 3057 the radiochemistry of radium. National Academy of Sciences - National Research Council. Nuclear Science Series
Scadden EM, Ballou NE (1960) NAS-NS 3009 the radiochemistry of molybdenum. National Academy of Sciences - National Research Council. Nuclear Science Series
Kujirai O, Yamada K, Fresenius KM (1991) Simultaneous determination of traces of impurities in high-purity molybdenum and molybdenum trioxide by coprecipitation and inductively coupled plasma-atomic emission spectrometry. J Anal Chem. doi:10.1007/BF00324398
Mogi F, Itoh K, Okamoto N, Narita M, Fujine M (1988) Determination of trace impurities in high-purity molybdenum and tungsten. Denki Seiko 59:263–270
Acknowledgements
We thank J.S. Choi and D. S. Leonard for ICP-MS measurements, and W.G. Kang and G.W. Kim for the HPGe measurements. This research was funded by the Institute for Basic Science (Korea) under project code IBS-R016-D1.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Gileva, O., Aryal, P., Karki, S. et al. Investigation of the molybdenum oxide purification for the AMoRE experiment. J Radioanal Nucl Chem 314, 1695–1700 (2017). https://doi.org/10.1007/s10967-017-5568-4
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-017-5568-4