
Abstract
The quality control of the method for tritium (3H) determination in water after electrolytic concentration by liquid scintillation counting measurements was improved considering more parameters in the uncertainty budget, besides the control charts to evaluate the electrolytic enrichment factor, the enrichment parameter and blank samples. The quality control upgrade of the method and the participation in proficiency tests allow its optimization. This methodology was applied to the determination of 3H activity concentrations in waters from different origins: surface, rain and drinking waters.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tritium (3H) is present in the environment because of natural and anthropogenic sources. It is a pure beta emitter (\(E_{ \hbox{max} } = 18.591 \pm 0.001 \;{\text{keV}}\)) with a half-life of \(T = 12.312 \pm 0.025 \;{\text{years}}\) [1, 2] and is produced in the atmosphere by nuclear reactions between fast neutrons that result from cosmic radiation and nitrogen atoms. Tritium levels in environmental samples increased between 1945 and 1963 due to nuclear testing. Since 1963, the activity concentration in 3H has been decreasing being current levels close to the values from pre-atomic era [3, 4]. This isotope is nowadays used as a global transient tracer for studying dynamics of the hydrological cycle. In particular, environmental tritium is widely used in hydrology as an age indicator for young groundwaters. With decreasing levels of environmental tritium, there is a need for sensitive measurement techniques capable of quantifying these levels with sufficient accuracy [5].
The ISO Standard 9698:2010 [6] is a guideline that specifies the conditions to perform the determination of 3H in waters using the direct measurement. When the need of lower detection limits (DL) is required, a solution is to perform the electrolytic concentration of the sample, the so called electrolytic enrichment. This procedure is carried out starting with the water sample purification by distillation, followed by the addition of sodium peroxide (Na2O2) before the electrolysis in direct current. After concentration of the sample, another distillation with PbCl2 is performed before liquid scintillation counting (LSC) in low-level mode [4].
To improve the quality control for the determination of tritium in waters using electrolytic enrichment, it became necessary to reevaluate the uncertainty budget considering more parameters [counting efficiency, 3H decay constant, weighings, total electrical charge, tritium enrichment factor (Z) and the enrichment parameter (P)] [7] since previous calculations only took into account the count rate. To check the electrolytic system performance, P and Z parameters and blank samples, are evaluated in each batch using Shewhart charts [8].
The laboratory takes part regularly in proficiency tests organized by international organizations such as European Commission, IAEA, Consejo de Seguridad Nuclear (CSN, Spain), etc. With the results obtained in these tests, it was possible to validate the accuracy of the method.
Theory
The methodology for 3H analysis with electrolytic enrichment is based on the ISO Standard 9698:2010 [5] and the required quality control was established according to ISO Standard 17025:2005 [9]. The method consists of four steps: sample treatment, electrolytic enrichment in direct current, sample neutralization and measurement by LSC in low-level mode [4], with previous calibration of the system using 3H standards [3].
The internal quality control comprises the samples storage (where the temperature and the tightness of the samples containers are checked), the sample’s and standards preparation by weighing in calibrated analytical balances and the verification of conductivity and pH of distillated samples. Besides that, the temperature and parameters of the electrolytic system are controlled and equipment performance tests are carried out before the sample measurements. To perform the external quality control, the laboratory participates regularly in proficiency testing schemes.
The sample activity concentration is calculated using the following Eq. (1), adapted from [5, 7]:
where C A is the activity concentration (Bq L−1), n the net count rate (cps), ε is the efficiency, S the sample amount (kg) and D is the decay constant.The combined standard uncertainty, as defined in the ISO/IEC guide to the expression of uncertainty in measurement [10], is given by the following expression (2):
Experimental
Methodology
Water samples are purified by vacuum distillation before measurement in order to remove any impurities, to reduce quenching, and to prevent the introduction of other interfering radionuclides. Distillation is performed by adding 0.5 g of Na2S2O3 and 1 g of Na2CO3 to a volume of about 500 mL [4]. The first milliliters of each sample are despised and 250 mL are preserved in borosilicate flasks until a minimum set of eight samples are completed.
During routine operation of the electrolytic enrichment system, each electrolytic run (14 cells with 250 g capacity each, connected by consecutive binding) usually consists of eight samples, three spike waters and three tritium-free waters to check the contamination level. For each run, three cells (different in each consecutive run) are filled with spike water (prepared from a tritiated (HTO) certified solution, Analytics) to determine the actual tritium enrichment through parameters P and Z [5]. All the cells are in a water bath (Valentim Lda) with about 220 L of coolant connected to a compressor BITZER CBS 33 V1.
This procedure is carried out by adding 1 g of sodium peroxide (Na2O2) to each cell (the sodium hydroxide formed is used as electrolyte). In each run the system is cooled to a temperature of about 2 °C and each electrolytic cell is submitted to an electrical charge until the reduction of the sample to a volume of about 15 g. The total electrical charge applied with a direct current source in a typical enrichment run is about 650 Ah, measured with an electronic ampere/hour meter (precision ±3 %). These equipments were produced in the electronic workshop at Nuclear and Technological Campus (CTN).
Afterwards the sample is neutralized by adding lead chloride (PbCl2) and distilled again in order to separate the lead oxide (PbO) and other impurities from the water [4].
To transfer water to the counting vials an analytical calibrated balance is used. An aliquot of the distillate (~8 g) is mixed with 12 g of Ultima Gold LLT™ scintillation cocktail in High Performance Glass Vials™, from PerkinElmer, borosilicate glass with low potassium content [3, 4]. Blank samples are prepared in the same conditions using distilled water (conductivity ~5.50 μS cm−1) without tritium content. Before counting, the glass vials are cleaned with an anti-static cloth and the samples are stabilized in the dark for about 8 h to minimize luminescence effects and reach thermal equilibrium. The measurements are performed in a Tri-Carb 3170 TR/SL (PerkinElmer) liquid scintillation counter in low-level mode [3] with the window for tritium set between 1 and 6 keV. The counting is performed for one cycle of 300 min in routine analysis, resulting in a DL of ~0.40 Bq L−1 (calculated according to [11]) with an efficiency of about 20 % and a background of ~0.03 cps.
The recovery of each cell and the system performance are obtained through Z and P parameters, respectively. For each run, these parameters are calculated and evaluated using control charts [8] with well-defined limits.
Quality improvement
The most important aspect of the quality of any method and respective result is the reliability of its associated statement of uncertainty, which must be an integral part of every analytical result. Several factors are involved in the methodology that may contribute to the observed variability. In this sense, in the estimation of the uncertainty budget, all sources of variability must be properly considered. Since 2014, besides count rate, already included in the uncertainty budget of the result, this calculation was improved by introducing parameters following Ref. [7], such as: counting efficiency, 3H decay constant, weighings, total electrical charge, Z and P parameters. After evaluating the contribution of the different components, it is possible to realize that the count rate, the sample amount (mainly the Z parameter) and the efficiency are the most relevant contributors to the uncertainty budget.
The counting efficiency is evaluated through quench curve instead of assumption of constant quench, like in Refs. [5, 7]. The results are provided by homemade software (internal procedure).
Equipment calibration
In order to perform the calibration of the liquid scintillation counter, 10 standard samples with an activity concentration of about 200 Bq were prepared (at a handling of radioactive sources laboratory) from a 3H certificate standard solution (Analytics) with a activity concentration of 411 Bq g−1 (August 2006). The samples were subjected to different degrees of quenching with the addition of different amounts of CCl4 (Merck) ranging from 0 to 20 μL to each standard sample (0, 2.0, 4.0, 6.0, 8.0, 10, 12, 14, 16, 18 and 20 μL, respectively) [3].The efficiency is obtained from the fitting of measurement data from the ten standard samples and it is evaluated according to (3),
where tSIE is the transformed spectral index of external standard. Figure 1 represents the calibration curve in function of the tSIE. The linear function is given by \(\varepsilon = \left( {8.53 \pm 0.32} \right)10^{ - 2} + (2.716 \pm 0.082)10^{ - 4} \times {\text{tSIE}}\). For usual quench samples (tSIE ~420) the efficiency of about \(0.200 \pm 0.005\) is achieved.

Calibration curve of counting efficiency as a function of quench parameter tSIE
Blanks and detection limit
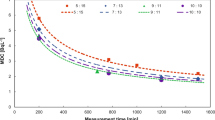
A batch of 10 blank samples is prepared for each calibration in order to calculate the average blank count rate and the limits for blanks control chart. This value is also used to estimate the DL. In routine measurements, a DL of about 0.40 Bq L−1 is achieved for 8 mL of enriched sample (obtained from the reduction of 250 mL to 25 mL of the initial sample) and a counting efficiency of 20 % for 300 min counting time (Fig. 2).

Detection limit (Bq L−1) versus counting time (h)
Calculations
The sample amount is evaluated by (4),
where m is the transferred sample to the scintillation vial (g), ρ is the water density (g cm−3) and Z smp is the electrolytic enrichment factor of the sample. To evaluate the last one, the initial (m 0) and final (m f) amount of the sample are weighted and Z smp is calculated according to (5):
where Ep is the enrichment parameter, Q is the total electrical charge (Ah) and F is the Faraday constant.
The decay constant is calculated for the time interval between the reference date and the starting of the measurement according to (6):
where T 1/2 (3H) is tritium half-life (s).
To evaluate the uncertainty budget related to these parameters, the uncertainties of the net count rate (7), the efficiency (8), the sample amount (9),the decay constant (10) and the electrolytic enrichment factor of the sample (11) are considered [5, 7, 10]:
where g is the gross count rate and b is the blank count rate. \(u\left( {E_{\text{P}} } \right)\) is calculated from the standard deviation of historical data.
In order to control the electrolytic process in each run, control charts to evaluate P, Z and blank samples are built and presented in Figs. 3, 4, and 5. The central line represents the mean value (\(\overline{X}\)), obtained with the historical data since the last calibration. The warning lines and the action lines are set at \(\overline{X} \pm 2\sigma\) and \(\overline{X} \pm 3\sigma\), respectively. If three values in a row fall between the warning lines and action lines, the process will be stopped and investigated. The same happens when one value fall outside the action lines.

Shewhart chart for P parameter with warning and action levels (Dec. 2014–Jul. 2016)

Shewhart chart for Z factor with warning and action levels (Dec. 2014–Jul. 2016)

Shewhart chart for blank samples with warning and action levels (Jan. 2015–Jul. 2016)
Results and discussion
As can be seen in the control chart, Fig. 3, the P parameter data are under statistical control with a mean value of 0.85, which is similar to the ones reported elsewhere [12, 13]. In Fig. 4 it is possible to observe that Z parameter values are lower than those reported by other authors [5, 12]. This could be related to the current inability of the system to concentrate the sample until the amount of 15 g. The count rate of the blank sample (Fig. 5) must be within the limits defined by the control chart, if not, it is necessary to prepare and measure a new blank sample.
Since 2014, following the improvement of the uncertainty budget calculation, the laboratory took part in two proficiency tests [14, 15]. The obtained results are presented in Table 1. It can be observed that in both cases the results were good (acceptable and satisfactory, respectively), according to the criteria established by the organizers and the laboratory (bias, uncertainty and Z-Score for results presented in [14] and Z-score for the ones in [15]). The results allowed the validation of the method accuracy. In addition, with a better caracterization of the method it was possible to decrease the DL comparatively to the value obtained before 2014 (0.8 Bq L−1) allowing the quantification of low activity concentration values as it can be observed for the sample IARMA-006 Level B.
This methodology was applied to water samples from different origins: surface, rain and waters intended for human consumption (drinking water), analyzed in the framework of the national environmental radioactivity monitoring programme. Table 2 shows the results obtained for 3H activity concentrations in waters collected monthly during the year 2015, in Tejo River (V. V. Ródão), Castelo Branco and Lisboa. The highest values, ranging from 0.94 ± 0.29 to 17.3 ± 1.4 Bq L−1, were observed for the surface waters. For rain waters the values were very low and in general below the DL (around 0.40 Bq L−1). Regarding drinking water, the mensurable values are situated between 0.54 ± 0.20 and 2.40 ± 0.37 Bq L−1. These values are far from the parametric value (around 2 orders of magnitude), 100 Bq L−1 established in the European Directive for water intend for human consumption [16]. The improvements introduced in the method allow a better characterization of the 3H activity concentrations mainly in rain and drinking waters which values are usually very low. Nowadays, it is possible to quantify the 3H activity concentrations in a major number of samples than before [4].
Conclusions
The method for the determination of tritium in water using electrolytic enrichment was improved with the reevaluation of the uncertainty budget, better characterization of DL and with the evaluation of counting efficiency through quench curve instead of assumption of constant quench. With the use of Shewhart charts it was possible to evaluate P and Z parameters and to control the cells in each run regarding blank samples. The methodology is under control since the mean values observed in each parameter lie between \(\overline{X} \pm 3\sigma\). However, some improvements should be introduced mainly concerning differences in the cells behaviour in each run. The accuracy was validated in dead water with different tritium concentrations and in a seawater matrix. The method was applied to waters from different origins, surface, rain and drinking waters being the highest 3H activity concentrations values obtained in surface waters. The future challenges are to improve P and Z parameters, giving primacy to the latter.
References
Tables of evaluated data. Laboratoire National Henri Becquerel. http://www.nucleide.org/DDEP_WG/DDEPdata.htm. Accessed April 2017
Bé MM, Chisté V, Dulieu E, Browne E, Baglin C, Chechev V, Kuzmenco N, Helmer R, Kondev F, MacMahon D, Lee KB (2006) Table of radionuclides (Vol. 3 – A = 3 to 244). Monographie BIPM-5. Pavillon de Breteuil, Sèvres, France
Gomes AR, Abrantes J, Libânio A, Madruga MJ, Reis M (2011) Improvement of the quality assurance in tritium analyses in waters by LSC. In: Cassette P (ed) LSC 2010, Advances in liquid scintillation spectrometry. Radiocarbon. ISBN: 978-0- 9638314-7-7. Arizona, USA, pp 211–218
Madruga MJ, Sequeira MM, Gomes AR (2009) Determination of tritium in waters by liquid scintillation counting. In: Eikenberg J (ed) LSC 2008, International conference on advances in liquid scintillation spectrometry. Radiocarbon. ISBN: 978-0-9638314-6-0. Arizona, USA, pp 353–359
Gröning M, Rozanski K (2003) Accreditation Qual Assur 8:359. doi:10.1007/s00769-003-0631-y
ISO Standard 9698:2010, Water quality—determination of tritium activity concentration—liquid scintillation counting method, 2nd edn
Quantifying uncertainty in nuclear analytical measurements. IAEA. http://www-pub.iaea.org/MTCD/publications/PDF/te_1401_web.pdf, pp. 195–217. Accessed April 2017
Miller JN, Miller JC (2010) Statistics and chemometrics for analytical chemistry. Pearson Education Limited, Harlow
ISO/IEC Standard 17025:2005—General requirements for the competence of testing and calibration laboratories, 2nd edn
Evaluation of measurement data—guide to the expression of uncertainty in measurement. JCGM 100:2008. http://www.iso.org/sites/JCGM/GUM/JCGM100/C045315e-html/C045315e.html?csnumber=50461. Accessed April 2017
ISO Standard 11929:2010, Determination of the characteristic limits (decision threshold, detection limit and limits of the confidence interval) for measurements of ionizing radiation. Fundamentals and application, 1st edn
Logar JK, Glavic-Cindro D (2009) Establishment of low-level tritium laboratory. In: Eikenberg J (ed) LSC 2008, International conference on advances in liquid scintillation spectrometry. Radiocarbon. ISBN: 978-0-9638314-6-0. Arizona, USA, pp 241–249
Baeza A, Garcia E, Miró C (1999) A procedure for the determination of very low activity levels of tritium in water samples. J Radioanal Nucl Chem 241(1):93–100
IARMA ETRIT-PT-2014 (2014) IARMA proficiency test on the determination of tritium in water at environmental levels
Trinidad JA, Gascó C, Llauradó M (2016) Evaluación de la Intercomparación CSN/Ciemat-2015 entre Laboratorios Nacionales de Radiactividad Ambiental (Agua)
European Directive 2013/51 EURATOM of 22 October 2013-laying down requirements for the protection of the health of the general public with regard to radioactive substances in water intended for human consumption. OJEU L296/12, 7.11.2013
Acknowledgements
The C2TN authors gratefully acknowledge the Fundação para a Ciência e Tecnologia (FCT) support through the UID/Multi/04349/2013 Project.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Gomes, A.R., Abrantes, J., Libânio, A. et al. Determination of tritium in water using electrolytic enrichment: methodology improvements. J Radioanal Nucl Chem 314, 669–674 (2017). https://doi.org/10.1007/s10967-017-5456-y
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-017-5456-y