
Abstract
The application of instrumental neutron activation analysis for the determination of long-lived rare earth elements (REE) in rock samples is considered in this work. Two different methods are statistically compared: the well established external standard method carried out using standard reference materials, and the internal standard method (ISM), using Fe, determined through X-ray fluorescence analysis, as an element-comparator. The ISM proved to be the more precise method for a wide range of REE contents and can be recommended for routine practice.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mineral resources analysis for individual rare earth element (REE) content is still a difficult task in the field of elemental analysis which can only be solved by spectroscopic (instrumental) methods. Recent reviews of spectroscopic techniques revealed the most common methods being used at present for geological sample analysis for REE content are inductively coupled plasma atomic emission spectrometry and mass spectrometry (ICP-AES and ICP-MS), X-ray fluorescence (XRF) analysis, and instrumental neutron activation analysis (INAA) [1, 2]. The basic restriction of the first two methods is that only solutions can be analyzed. Complete sample dissolution is required and this may be a problem in the case of REE determination since, like the other lithophilic elements, they are associated with silicate rocks. The main disadvantages of ICP-AES are the poor sensitivity of REE analysis, a substantial matrix effect, the intricacy of the REE individual spectra and spectral interferences. To avoid most of these disadvantages, efficient pre-concentration and separation procedures need to be employed [3, 4]. The ICP-MS method is much more appropriate for REE analysis, but it is also subject to spectral interferences including difficult to correct for polyatomic ions which the lanthanides usually form in plasma. The typically low content of REEs in rock means careful sample preparation is essential to avoid analyte loss and/or solution contamination, making the entire procedure a crucial part of the analysis [5, 6]. As a result, the ICP-MS technique becomes the most expensive one for REE determination [1].
Unlike ICP-AES and ICP-MS, XRF is commonly used for nondestructive analysis, however in the case of REE measurement it suffers from insufficient sensitivity both in the energy dispersive (isotope excitation of K-series [7]) and the wave dispersive (X-ray tube excitation of L-series [8]) variants. Only some light lanthanides (La, Ce, Pr and Nd) can be analyzed by XRF due to their higher average crustal contents. To determine heavier lanthanide content, a time- and reagent-consuming procedure of sample decomposition followed by chemical separation from the major matrix elements and concentration on a target should be applied [9]. Spectral interferences and a substantial matrix effect present a serious problem in XRF, so high resolution spectrometers and sophisticated algorithms must be used, increasing the complexity of the whole analysis.
In contrast to XRF, neutron activation analysis is “physically fully described and understood”, and that is considered its main strength [10]. Compared to the other methods, rock analysis for REE content is where INAA shows its best advantages. The most important of these advantages are nondestructive sample treatment and hence a virtual absence of analytical blank, high sensitivity making possible all REE measurements at levels much lower than average crustal contents, relative freedom from the matrix effect and few spectral interferences (mainly caused by the U fission products) [11]. Repeated comparison of the precision of INAA and ICP-MS methods with the help of geological reference materials showed the former to give, in general, better results, especially for the middle and heavy REEs [12, 13]. INAA is characterized by a relatively low price for REE determination [1] due to the wide-spread accessibility of research reactors around the world. All these advantages promote the broad application of INAA to geological sample analysis for major and trace elements [14], noble metals [15], and REE [16–22] content.
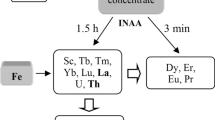
Some approaches to the methods classification must be mentioned. Both the relative and the non-relative (direct) methods of concentration standardization are used in INAA [11]. The latter comprises the absolute and the single comparator methods (SCM). General definitions of the relative and direct methods are cited by Greenberg [23]. If one (the SCM) or more (the relative method) comparator elements are separated from an analyzed sample, then both methods use “the external standard method” of calibration (external standardization) which is most often applied in analytical chemistry. “The internal standard method” (ISM) is mainly used for liquid sample analysis and is based on the known content of a comparator element in every sample. A physical method must allow the calculation of other analyzed elements, and INAA is one such method. In this case, internal standardization is an additional way of calibration in the SCM, but it cannot be applied with the relative method. The interrelation between INAA methods varying with concentration standardization and the method of calibration is presented in Fig. 1. Grey capsules denote reference material samples and the black capsule denotes a single element comparator.

Interrelation between two ways of concentration standardization (a, b) and two ways of calibration (b, c) in INAA
Methodologically rather simple and hence widely used, relative INAA needs the rock reference materials certified for REE content (CRMs)Footnote 1 in order to be used both as calibration standards and for implementation of the analytical quality control procedures. The main disadvantages of the relative method are additional time consumption, considerable cost and limited choice of high-quality CRMs with the uncertainties associated to REE not exceeding 8–10 % (e.g. by the Institute for Reference Materials and Measurements (IRMM), Belgium). More accessible CRMs do not guarantee adequate precision (e.g. fixed in [24]) due to high uncertainties of REE certified values. Gamma-ray intensity corrections for spectral interferences and U fission products must often be made for the CRMs too, since U, Th and other high element contents compete with REEs. Additional analytical uncertainty may arise from reference material heterogeneity since the CRM subsamples taken for irradiation are frequently less than “the minimum sample intake” recommended by the certificate. Lastly, analytical results depend on the CRM manufacturer and can vary more than 10 % without any apparent reason.
The single comparator method, including k 0-INAA, removes the majority of the disadvantages of the relative method resulting from the use of CRMs, since the CRMs are only used for analytical quality control. The basic drawback of the SCM is a consequence of the comparator method itself. Since generally the comparator material and the samples present different features, the latter should be considered when applying some corrections. In separate cases, the SCM can result in lower performance if compared to the relative method due to the effect of thermal and epithermal neutron self-shielding. The SCM method is commonly characterized by a more sophisticated calculation since it needs several parameters to be applied [25, 26].
If the SCM uses the internal standard method (an internal comparator) it eliminates its main drawback by taking into account the effects of different geometries and the neutron self-shielding in every sample. In addition, the neutron flux gradient inside the irradiation container should be considered: if a source of mostly thermal neutrons is used for irradiation, then the values of epithermal to thermal neutron flux ratio, 1/f, in every sample can also be estimated. However 1/f is assumed not to vary within an irradiation container at all, not even from day to day [25]. Hence, when similar samples are irradiated in the same channel position, the 1/f value can be considered as a constant and there is no need to correct for it. The mass fraction of any element of interest can be found using the known content of a single element comparator only.
There are different ways to determine the internal comparator content of a sample. The standard k 0-method with Al–Au foil or wire as a flux monitor is applied most often [27, 28]. For better accuracy and for bulk or irregular geometry sample analysis, the INAA relative method is used [29–32]. The internal comparator content can be also found by an independent method. Matrix or major elements are usually selected to implement internal standardization: Ca for compositional characterization of cement samples [31], Fe to measure element contents of meteorites [29], uranium ore [30] and high-purity aluminum-clad samples [32], and Na for large dross sample analysis for Au and Ag content [33]. Sometimes other elements can be used, such as Sc for the elemental analysis of ancient pottery samples [34], Rb to analyze plant leave samples [27], and La for INAA ISM verification by means of Soil-7 CRM [28].
In this paper the precision of INAA for long-lived REE using the relative method (Fig. 1a) and using the internal standard method (Fig. 1c) with XRF as an independent analytical technique is compared for a wide range of REE content in geological samples covering several orders of magnitude.
Theory
Any element content of the analyzed sample C a (%) by the internal standard method can be calculated using the equation of the single comparator method of standardization [35, 36] (lower case indices a and c mean an analyzed element and the comparator, respectively):
where C c is the element comparator content of the sample (%), J is the full-energy peak count rate of the corresponding radionuclide analytical gamma-ray (cps), ε(E) is the relative detection efficiency of the analytical gamma-ray (%), Q 0 is the resonance integral I 0 (cm2) to thermal neutron cross-section \(\sigma_{0}\) (cm2) ratio, f is the thermal to epithermal neutron flux ratio, S and D are, respectively, saturation and decay factors, C is the correction for measurement time, G is the correction factor for neutron self-shielding by the sample, F is the correction factor for analytical gamma-ray self-absorption by the sample, and the k-factor is composed of nuclear constant product:
where θ is isotopic abundance (%), \(P_{\gamma }^{{}}\) is the yield of the analytical gamma-line (%), M is the atomic mass (Da). The empirical correction factor K a,c compensates for a bias which may be caused by the errors of the detector calibration for relative detection efficiency.
In the case of the long-lived radionuclide analysis, counting correction C is always <1 % and can be neglected. Correction factor G is very close to unity in most samples and also can be ignored [36]. If the internal comparator is determined by an independent technique, INAA ISM may be used to analyze materials suffering from thermal neutron self-shielding since it affects all the elements in the sample to the same degree [27]. Even in the case of REE high content analysis, self-shielding can be usually kept to a negligible value (no more than 5 %) by diminishing the sample mass. Correspondingly, the F factor must be taken into account only in the case of large sample analysis [37] or a heavy matrix [38], and can be neglected when irradiating small samples with a common Al–Si matrix.
To evaluate the 1/f ratio in rock samples, the content of at least two easily detectable elements with respectively a low and high Q 0 value must be known. Considering Fe as the element with a low Q 0 value, the second element could be Rb, Sr, Ba, Cs, Th or U, depending on its content. The values of 1/f can be calculated solving the system of two elements using Eq. (1). Then, keeping the ordinary conditions of rock analysis such that the correction factors mentioned above can be neglected, the epithermal to thermal neutron flux ratio equals to \((Q_{0,1} << Q_{0,2} )\):
where
lower indices in both Eq. 3 are valid. This method is effective where the neutron flux is not highly thermalized. If the neutron flux is highly thermalized then the uncertainty of the calculated value of 1/f would be too high; in this case the cadmium ratio method should be used.
Experimental
Following the approach above, it was decided to use 58Fe as the internal standard for the determination of REE in geological samples. Iron was chosen as it is widely spread in all rock types with high crustal abundance, and the radionuclide 59Fe is characterized by suitably low value of resonance integral to thermal neutron cross-section ratio and a rather long half-life time. The Fe content of the rock samples was determined by an independent method, XRF analysis, using a portable energy dispersive X-ray spectrometer RLP-21T (Republic of Kazakhstan). The ascribed uncertainties of such measurements are 2–8 % [39], comparable with the uncertainties associated with REE in the high quality CRMs. The mass of an ordinary rock sample (Al–Si matrix) measured with RLP-21T is 5–7 g, much greater than if INAA is applied to analyze the same material. The larger sample mass is an important advantage of XRF, since the representativeness of the small geological material subsamples typically used for trace element analysis may be questionable [40]. Then, using the Fe content measured by XRF as the internal standard, the reproducibility of rock analysis for REE content should be improved compared with the INAA relative method. Moreover, XRF complements INAA well in mineral resources exploration, enabling the Fe content of rock samples to always be available [41, 42].
To assess 1/f and K a,Fe values, the archive of INAA measurements relative to different multi-element CRMs carried out over an eighteen month period was consulted. Correction coefficients K a,Fe used for the determination of REE content with respect to Fe as the internal standard were evaluated, based on a total number of 50–70 measurements; 5–10 different types of CRMs—rocks, soil, flying ash, and bottom sediments were used. Some CRMs were rejected since they resulted in K a,Fe values for certain REEs being incompatible with other ones within a reasonable discrepancy (10 %), or with insufficient reproducibility of separate K a,Fe estimations. Apart from low gamma-ray intensities, the discordance was related to cases of high uncertainty in the REE certified values and to cases of significant spectral interferences. Substantial scattering of individual K a,Fe values was marked for the CRMs with poor small subsample representativeness. Both cases were mentioned above as drawbacks to the relative method.
The relative bias of REE content measurement by INAA ISM was evaluated using CRMs certified only for REE contents, but not for Fe content: BCR-667 (estuarine sediment, IRMM), CC-690 (calcareous soil, ERM), and SL-3 (river sediments, IAEA). The Fe content of these CRMs was measured using an RLP-21T X-ray spectrometer. The samples were prepared, irradiated and measured as described below.
The RLP-21T spectrometer by JSC “Physicist”, Almaty, was designed to analyze powdered geological samples (rocks, ores, soil, sediments) and ore concentrates. In order to get maximum accuracy, the elemental content of the samples was calculated using a reference-free modified method of the fundamental parameters. The spectrometer is enrolled in the State Register of Measuring Devices, and the certified analytical technique is registered by the National Body for Certification of Kazakhstan. The accuracy of calculations by the spectrometer software was repeatedly confirmed with the help of appropriate, different CRMs. E.g., the Fe content of rock reference materials is determined with a systematic error of no more than 4–5 %.
To evaluate the relative standard deviation of INAA for long-lived REE by the ISM, the archive data on the results of independent measurements of rock samples by the relative standardization method were used. These data were obtained in the course of certified analytical technique [43] elaboration and corresponded to all concentration ranges of REE analysis by this technique. The data were replenished later with additional measurements to provide some content intervals with minimum numbers of replicates.
Most of the rock samples used for the investigation were collected from Kundybai REE deposit, Northern Kazakhstan, and prepared for instrumental analysis according to the routine technique with a particle size <0.074 mm. The CRMs used were certified for REE content and obtained from different manufacturers such as IAEA, IRMM, China, Poland, and the Russian Federation.
50–200 mg of rock samples, depending on the expected REE content, and 100 mg of CRMs were used. Samples were sealed in small double polyethylene bags, packed in Al foil and placed in an irradiation container. Up to 23 bags were irradiated at the same time for 1–2 h in position № 3 inside “wet” channel № 8–9 of the research reactor WWR-K with a typical neutron flux density 1013 cm−2 s−1. No flux monitors were used because the relative method was applied.
Irradiated samples were measured twice: first after 7 days of the decay time for the determination of La, Nd, Sm, Ho, Yb, Lu and secondly about 30 days after the irradiation for the determination of Sc, Ce, Eu, Gd, Tb, and Tm. Both measurements were performed using an extended-range HPGe detector GX5019 with a relative efficiency of 50 % and an energy resolution of 1.86 keV at the 1332 keV peak of 60Co, connected to a Canberra multi-channel analyzer DSA-1000. Detector calibration for relative detection efficiency was made with the help of a multi-gamma ray standard (152Eu, 154Eu, 155Eu) and an isotopic source 133Ba, both by Canberra. Spectra collection and subsequent treatment were carried out by the special software developed in the INP to provide gamma-ray spectrometric analysis. The coefficients to 239Np full-energy peak intensities to correct La, Ce, Nd and Ho analytical line count rates for U fission products were calculated by counting a specially prepared sample made from a uranium standard solution by Perkin Elmer. No corrections for neutron self-shielding, gamma-ray self-absorption or true-coincidence effects were applied.
The main nuclear parameters of the analytical gamma-ray lines of the radionuclides used to calculate REE content and the interferences which were taken into account are presented in Table 1. U(n,f) means the same radionuclide as a U fission product, whereas 133Xe by the U fission reaction appears as a spectral interference. Some minor interferences in an ordinary rock matrix were regarded as inessential and neglected. The partly resolved peaks, where possible, were divided by the software.
Results and discussion
The epithermal to thermal neutron flux ratio and its time stability in the selected position of the irradiation channel were estimated using the 1/f values calculated according to Eq. (3) with the help of several CRMs irradiated at different times. Rb and Cs were chosen for the determination of 1/f since they presented the higher number of independent measurements (respectively 50 and 46) among the elements with a high Q 0 value. The determined 1/f values are presented in Fig. 2, where they are reported in chronological order. No time trends were revealed for either of them, as expected.

Calculated values of epithermal to thermal neutron flux ratio using respectively Fe-Rb (a) and (Fe-Cs) (b) determined in the CRMs
As they were composed of independent data, the ranges can be treated as stochastic variables with mathematical statistics methods applicable to them. First, the strict nonparametric Mann–Whitney U-test for independent samples [44] insensitive to variable distributions was employed to compare the two ranges. Since the calculated value of the standard normal variable (SNV) \(\hat{z} = 0.87\) did not exceed its critical value \(z_{0.05} = 1.96\) for the two-tailed test, it was concluded that these two datasets came from the same population and were combined. The nonparametric Kolmogorov–Smirnov test showed the distribution of the combined dataset did not contradict the Gaussian one (the calculated value of D-statistics \(\hat{D} = 0.0616\); the critical value \(D_{0.05} = 0.139\); n = 96). Thus the combined dataset parameters can be described in the terms of normal distribution, i.e. the 95 % confidence interval embraces two standard deviations σ around the mean. The epithermal to thermal neutron flux ratio in the selected irradiation position appeared to be: 1/f = 0.028 ± 0.004 (P = 0.95, σ = 0.002).
In spite of a great number of measurements, the uncertainty of this value is rather high, about 14 %. Hence, this approach apparently cannot be applied to evaluate 1/f in distant positions of the irradiation channels with a more thermalized neutron flux. In this case the cadmium ratio method must be used.
The evaluated neutron flux ratio made possible the estimation of relative bias values (trueness) of the REE content measurement of the selected CRMs: Δrel = (C INAA/C CRM – 1) × 100 %, where C INAA is the REE content measured by INAA using the internal standard method, and C CRM is the corresponding certified value. The rounded Δrel values are presented in Table 2. Blanks mean an absence of certified values.
The acceptable |Δrel| values of no more than 10 % supported further comparison of the precision of INAA for these REEs by the relative and internal standard method. Selected results of REE analysis in rock samples by the relative method were reinterpreted using the Fe contents of these samples obtained by XRF as the internal standard. Based on 5 replicas, 12 pairs of the mean REE values C m and the relative standard deviations s/C m characterizing reproducibility of each REE measured by both methods were calculated. The uncertainties of the single measurements derived by the relative method are fixed, [43], ranging within 12–22 % for all the REE presented in Table 2, with the exception of Gd and Ho (16–28 %). Thus, the relative uncertainties of C m values should be a factor of \(\sqrt 5\) lower, i.e. approximately 6–10 and 7–13 % correspondingly. Although it would be done for certified analytical techniques, there was not a systematic assessment made of REE measurement uncertainties by INAA ISM. However these values can be assumed not to exceed the ones derived by the relative method, since the internal standardization is specifically used to increase reproducibility and accuracy of a quantitative analysis [45].
The dependency of s/C m values on REE content approximated by nonlinear power law trends are plotted in Figs. 3, 4 in semi-logarithmic coordinates. These graphs show that all the trends corresponding to the ISM are located below the trends corresponding to the relative method. Such an order supposes reproducibility of the ISM to be better in all the cases, but due to the random character of the variables this must be proved by mathematical statistical methods.

Relative standard deviation values of light REE analysis by INAA: Sc (a), La (b), Ce (c), Nd (d), Sm (e), Eu (f)

Relative standard deviation values of heavy REE analysis by INAA: Gd (a), Tb (b), Ho (c), Tm (d), Yb (e), Lu (f)
If highly homogeneous powder or water samples are analyzed, the experimental dependence of s/C m on analyte content appears as a hyperbola-like descending curve. Such behavior was not shown for Sc, La, Eu, Yb analysis by the relative method and La, Nd, Yb analysis by the ISM; the empirical trends are slopeless or not sufficiently sloped, as could be expected. This effect was explained by the increasing heterogeneous REE distribution within the samples collected from the real REE deposit. It appears, in this case, the usual grinding to a particle size <0.074 mm was not sufficient to provide the necessary homogeneity in small samples.
To establish the quantitative distinction between trend behavior for every REE, two statistical tests were applied using different approaches. The first test is “comparison of one empirical curve with another after some interference in the process” [44] (“ECC”). It is based on the construction of the standard normal variable (SNV) \(\hat{z}\) in the following way:
where, for the case under discussion, y 1,i and y 2,i are the pairs of s/C m values for the same REE content (see Figs. 2, 3), n = 12, s 1 and s 2 are the standard deviations of y 1 and y 2 respectively. The test implies Gaussian distribution of the independent s/C m variables forming each trend, and this hypothesis was confirmed by effective nonparametric Shapiro–Wilk W-test applicable for restricted samples. As an alternative, the nonparametric Mann–Whitney U-test was applied to each of twelve pairs of s/C m stochastic samples to verify their belonging to the same general population.
The calculated SNV values for both approaches are presented in Table 3. Comparing them with the critical value \(z_{0.05} = 1.96\), a conclusion was drawn that for Gd, Ho and Lu the application of the relative method or the ISM do not present any difference in terms of analytical performance. This is not valid for the other REEs: in these cases the application of the ISM offers an advantage.
A further comparison of the ability of the INAA ISM and the relative methods to analyze the REE content of geological objects was made in terms of their precision assessment. The next three CRMs by Ore Research & Exploration, Australia, were used to that end: OREAS 24b (granodiorite), OREAS 100a (uranium-bearing multi-element reference material) and OREAS 463 (REE-Nb ore). Sample preparation and measurement were carried out basically as described above. Three replicas of each CRM were irradiated simultaneously for 2 h:100 mg of the first two and 10 mg of OREAS 463. The subsample masses of the third CRM were substantially diminished to reduce the neutron self-shielding effect (due to high REE content) to a negligible value.
All measurements were carried out using the main GX5019 detector and the additional GLP36360 planar detector (energy resolution is 585 eV at 122 keV peak of 57Co) by ORTEC. GLP36360 was used for Gd analysis since it partly resolves the 103.18 keV analytical peak of 153Gd from the 103.86 keV interference line of 233Pa. GLP36360 does not discriminate between the energies of 166Ho (80.57 keV) and 133Xe (81.00 keV) lines, but it resolves the sum peak and thus made Ho analysis in OREAS 100a more reliable. In this case 233Pa (311.9 keV) was used as the internal standard instead of 59Fe, since the count rate of the 192.3 keV line of 59Fe (quantum yield is 3.1 %) measured by the planar detector was too low. The Th content of OREAS 100a was determined by INAA ISM with the help of GX5019 and using Fe as the internal standard. Thus, to analyze Ho in OREAS 100a the ISM was applied twice. The same detector was used to analyze Ho in OREAS 24b due to insufficient count rate by the planar detector. Tm and Ho were not determined in Oreas 463 because of severe overlapping by 182Ta and the corresponding absence of the analytical signal against the background.
The REE content of the OREAS samples by the relative method was calculated using the following CRMs: GBW-07110 (trachyte andesite) and GBW-07159 (rare earth ore), both from China.
The OREAS CRMs’ certified values (±1 standard deviation) of long-lived REEs, Th and U along with their measured mean values by INAA ISM and relative methods calculated using three replicas are presented in Tables 4, 5 and 6. A blank means the absence of a certified value. The Sc content of OREAS 463 is an indicative one.
A quantitative comparison of precision between the two INAA methods versus the certified values showed a noticeable advantage using the INAA internal standard method for REE analysis in geological samples of different composition. Together with the demonstrated higher reproducibility of small rock sample analysis, INAA, using Fe as the internal standard with the content determined by an up-to-date XRF technique, can be successfully used for long-lived REE analysis in routine practice.
Conclusions
The experimentally-proven stability of neutron spectra in the same irradiation position of the research reactor WWR-K enabled the application of the simplified approach to INAA of geological samples for rare earth element determination using the internal standard method. Taking account of its nuclear properties and wide abundance in rocks and minerals, Fe looks to be the most suitable internal comparator. An independent up-to-date instrumental method such as energy dispersive XRF can be a convenient means of Fe content determination. The absence of a range of drawbacks inherent in the relative method is the most attractive feature of INAA ISM. Its main disadvantage is the same as for SCM, in that they are both strictly bound to experimental counting conditions.
The estimated precision, high reproducibility of small sample analysis, and other advantages, enables INAA ISM to be recommended for routine analysis of a large series of similar rock types for REE content.
Notes
Include SRMs, ERMs or any other national trademarks of certified reference materials.
References
Gorbatenko AA, Revina EI (2015) A review of instrumental methods for determination of rare earth elements. Inorg Mater 51:1375–1388
Zawisza B, Pytlakowska K, Feist B et al (2011) Determination of rare earth elements by spectroscopic techniques: a review. J Anal At Spectrom 26:2373–2390
Rajendran J, Thampi PK, Balasubramanian G (2006) Determination of rare earth elements in garnet minerals, geological materials by inductively coupled plasma-atomic emission spectral and mass spectral analysis. Anal Lett 39:2297–2306
Premadas A (2003) Cation exchange chromatographic group separation and ICP-AES determination of rare earth elements and yttrium in refractory minerals zircon, ilmenite, rutile, columbite-tantalite, garnet, and silliminite. At Spectrosc 24:149–158
Frederico GP, Rainério EJ, Tatiana DS (2012) Sample preparation for determination of rare earth elements in geological samples by ICP-MS: a critical review. Anal Lett 45:1537–1556
Fedyunina NN, Seregina IF, Bolshova MA et al (2012) Investigation of the efficiency of the sample pretreatment stage for the determination of the rare earth elements in rock samples by inductively coupled plasma mass spectrometry technique. Anal Chim Acta 713:97–102
Zuzaan P, Gansukh N, Bolortuya D (2010) Radionuclide induced energy dispersive X-ray fluorescence for the determination of La, Ce, Pr and Nd and their content sums in the rare-earth ores. X-ray Spectrom 39:52–56
Khudonogova EV, Suvorova DS, Revenko AG (2015) X-ray fluorescence determination of Cs, Ba, La, Ce, and Nd concentrations in rocks of various compositions. Anal Kontrol 19:347–356
Orescanin V, Mikelic L, Roje V et al (2006) Determination of lanthanides by source excited energy dispersive X-ray fluorescence (EDXRF) method after preconcentration with ammonium pyrrolidine dithiocarbamate (APDC). Anal Chim Acta 570:277–282
Bode P (2012) Opportunities for innovation in neutron activation analysis. J Radioanal Nucl Chem 291:275–280
IAEA-TECDOC-1215 (2001) Use of research reactors for neutron activation analysis. International Atomic Energy Agency, Vienna
Papaefthymiou H, Papatheodorou G (2011) A comparison between INAA and ICP-MS for the determination of element concentrations in marine sediments. J Radioanal Nucl Chem 289:679–688
Dai Kin F, Prudencio I, Gouveia A et al (1999) Determination of rare earth elements in geological reference materials: a comparative study by INAA and ICP-MS. Geostand Newsl 23:47–58
Baccolo G, Clemenza M, Delmonte B et al (2015) Assessing the geochemical fingerprint of the 2010 Eyjafjallajokull tephra through instrumental neutron activation analysis: a trace element approach. J Radioanal Nucl Chem 306:429–435
Rodrıguez N, Yoho M, Landsberger S (2016) Determination of Ag, Au, Cu and Zn in ore samples from two Mexican mines by various thermal and epithermal NAA techniques. J Radioanal Nucl Chem 307:955–961
Lim JM, Jeong JH, Lee JH (2013) Instrumental neutron activation analysis of coal and its combustion residues from a power plant. J Radioanal Nucl Chem 298:201–208
Capannesi G, Rosada A, Manigrasso M et al (2012) Rare earth elements, thorium and uranium in ores of the North-Latium (Italy). J Radioanal Nucl Chem 291:163–168
El-Taher A (2010) Rare earth elements content in geological samples from eastern desert, Egypt, determined by instrumental neutron activation analysis. Appl Radiat Isot 68:1859–1863
Sabiha-Javied Waheed S, Siddique N et al (2010) Measurement of rare earths elements in kakul phosphorite deposits of Pakistan using instrumental neutron activation analysis. J Radioanal Nucl Chem 284:397–403
Rezaee Kh, Saion EB, Khalik Wood A (2010) Rare earth elements determination and distribution patterns in surface marine sediments of the South China Sea by INAA, Malaysia. J Radioanal Nucl Chem 283:823–829
El-Taher A (2007) Rare-earth elements in Egyptian granite by instrumental neutron activation analysis. Appl Radiat Isot 65:458–464
Ravisankar R, Manikandan E, Dheenathayalu M et al (2006) Determination and distribution of rare earth elements in beach rock samples using instrumental neutron activation analysis (INAA). Nucl Instrum Meth Phys Res Sect B 251:496–500
Greenberg RR (2008) Pushing the limits of NAA: accuracy, uncertainty and detection limits. J Radioanal Nucl Chem 278:231–240
Gusev S, Ryabev V, Vorobyov V, Masalovich N, Lubimova L (2004) OST 41-08-212-04 industrial standard. Quality management of analytical work. Error guidelines for chemical analysis of mineral resources and precision classification of laboratory analytical techniques. Published by FSMC VIMS, Moscow (in Russian)
The k0-IAEA Program Manual for Version 5.00, International Atomic Energy Agency. http://www-naweb.iaea.org/napc/iachem/K0-IAEAhelp.pdf. Accessed May 03 2016
Greenberg RR, Bode P, De Nadai Fernandes E A (2011) Neutron activation analysis: a primary method of measurement. Spectrochimica Acta Part B 66:193–241
Lin X, Henkelmann R (2004) The internal comparator method. Anal Bioanal Chem 379:210–217
Sudarshan K, Nair AGC, Goswami A (2003) A proposed k0 based methodology for neutron activation analysis of samples of non-standard geometry. J Radioanal Nucl Chem 256:93–98
Nair AGC, Acharya R, Reddy AVR et al (2004) Elemental composition of Jagannath meteorite by neutron activation analysis. Curr Sci 87:654–657
Tiwari S, Nair AGC, Acharya R et al (2007) Analysis of uranium bearing samples for rare earth and other elements by k0-based internal monostandard INAA method. J Nucl Radioch Sci 8:25–30
Sudarshan K, Tripathi R, Acharya R et al (2014) Application of k0-based internal mono-standard PGNAA for compositional characterization of cement samples. J Radioanal Nucl Chem 300:1075–1080
Shinde AD, Acharya R, Reddy AVR (2014) Trace element determination in high-purity aluminum clad samples by k0-based internal monostandard instrumental neutron activation analysis. J Radioanal Nucl Chem 299:1287–1292
Swain KK, Ajith Nicy, Acharya R et al (2012) Large sample neutron activation analysis of dross for gold and silver. J Radioanal Nucl Chem 294:319–322
Dasari KB, Acharya R, Swain KK et al (2010) Analysis of large and non-standard geometry samples of ancient potteries by internal monostandard neutron activation analysis using in situ detection efficiency. J Radioanal Nucl Chem 286:525–531
De Corte F (1987) The k0-standardization method. University of Gent, A move to the optimization of neutron activation analysis
Zs Revay, Kennedy G (2012) Application of the k0 method in neutron activation analysis and in prompt gamma activation analysis. Radiochim Acta 100:687–698
Menezes BC, Jacimovic R, Pereira C (2015) Spatial distribution of neutron flux in geological larger sample analysis at CDTN/CNEN, Brazil. J Radioanal Nucl Chem 306:611–616
Chilian C, Lacroix C (2014) Towards routine NAA of materials rich in heavy elements with iterative gamma-ray attenuation and neutron self-shielding calculations. J Radioanal Nucl Chem 300:547–552
Determination of element content of different material powder samples by EDXRF spectrometer with semiconductor detector RLP-21T. Analytical Technique KZ.07.00.01569-2012, Kazakhstan Institute of Metrology. http://kazinmetr.kz/si/index.php. Accessed May 03 2016
D’Agostino G, Bergamaschi L, Giordani L et al (2014) Use of instrumental neutron activation analysis to investigate the distribution of trace elements among subsamples of solid materials. Metrologia 51:1–6
Solodukhin V, Silachyov I, Poznyak V, et al. (2016) Development of the complex of nuclear-physical methods of analysis for geology and technology tasks in Kazakhstan. J Radioanal Nucl Chem. doi:10.1007/s10967-016-4764-y (accepted for publication)
Popelka-Filcoff RS, Robertson JD, Glascock MD et al (2007) Trace element characterization of ochre from geological sources. J Radioanal Nucl Chem 272:17–27
Determination of REE Lu, Tb, Tl, Eu, Ho, Yb, Sc, Sm, Gd, La, Nd, Ce, and Y content of geological samples by the complex of nuclear-physical methods. Analytical Technique KZ.07.00.02096-2014, Kazakhstan Institute of Metrology. http://kazinmetr.kz/si/index.php. Accessed May 03 2016
Sachs L (1972) Statistische Auswertungsmethoden. Springer-Verlag, New York
Lebedev A (2012) Comprehensive environmemtal mass spectrometry. ILM Publications, St Albans, Glendale
Acknowledgments
The author is greatly thankful to Dr John W. Bennett and Ms Gillian Blackburn, Australian Nuclear Science and Technology Organization, for valuable remarks and corrections in the course of manuscript preparation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Silachyov, I. Rare earths analysis of rock samples by instrumental neutron activation analysis, internal standard method. J Radioanal Nucl Chem 310, 573–582 (2016). https://doi.org/10.1007/s10967-016-4903-5
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-016-4903-5