
Abstract
Using polyurethane foams integrated with phase change materials (PCMs) that take cooperative advantages of heat insulation and heat storage capacity can meet the demand for thermal comfort and energy conservation purpose in the buildings. One-shot synthesis method, a cost-effective method, was used in this study for fabrication of PU-PCM composite foams. It is ascertained that thermal regulation capacity of composite foams is facilely tunable as latent heat storage properties of composite foams containing 10–20 wt.% n-octadecane as PCM ranged from 17.72 J.g−1 to 34.51 J.g−1. Inclusion of PCM resulted in increasing the cell size of composite foams accompanied with reduced closed-cell content compared with those of pristine PU foam. However, as the PCM fraction increased, the cell size of composite foams tended to decrease. Given the fact that the geometry of foam cells determines specific compressive properties, specific compressive strength and modulus of PU foam dropped from 3.89 ± 0.20 kPa.m3.kg−1 and 77.48 ± 5.72 kPa.m3.kg−1 to 2.67 ± 0.43 kPa.m3.kg−1 and 46.05 ± 9.86 kPa.m3.kg−1, respectively after incorporation of 10 wt.% n-octadecane. However, as the foam cell size reduced by increasing the PCM content, specific compressive strength and modulus were improved and reached 3.44 ± 0.30 kPa.m3.kg−1 and 65.50 ± 1.16 kPa.m3.kg−1, respectively for composite foam containing 20 wt.% n-octadecane which are comparable to those of PU foam. Additionally, the PCM leakage from this composite foam was less than others. This study suggests that by adjusting the PCM content, one-shot synthesized PU-PCM composite foams can provide not only reasonable thermal regulation properties and appropriate thermal reliability but also sufficient mechanical properties.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The building sector is account for 30–40% of the total world’s energy consumption as well as one-third of the global greenhouse gas emission [1]. Insulation materials can contribute to enhancement of the energy efficiency of building envelopes through reducing the heating and cooling energy consumption, providing thermal comfort, and decreasing the carbon emission [2,3,4,5,6]. The performance of an insulation is generally expressed in a static term as the thermal resistance which is a measurement of a material ability to oppose the flow of heat for a static temperature difference between two faces. To achieve higher thermal resistance, traditional insulation materials are used in thick or multiple layers resulting in a complex building details, adverse effect on net-to-gross floor area and heavier load bearing construction [2]. These traditional insulating materials can be replaced with the rigid polyurethane (PU) foam, an excellent thermal insulating material with low density, superior specific mechanical properties, chemical resistivity, and high anti corrosiveness [1, 7]. The PU foam is prepared by polymerization reaction between polyol and diisocyanate in presence of a proper amount of catalysts, surfactants, and blowing agents [8]. On the other hand, the static term, thermal resistance, can be extended to a dynamic term, the heat storage coefficient, expressing the resisting ability of the building envelope to a periodical heat flow wave which depends on the thermal conductivity, the volumetric heat capacity, and the seasonal heat flow wave [2]. Thermal energy storage (TES) systems as the new-efficient module with capability to storage thermal energy in the form of latent heat for later use have received considerable attention in over the last decade in order to increase the heat storage coefficient of insulation materials [2, 6]. Phase change materials (PCMs) also called the latent heat storage (LHS) are capable of storing or releasing energy in large quantities during a phase change within minor temperature variations [1, 9]. The PCM-incorporated PU foams can benefit from both thermal resistance and thermal energy storage [1, 6, 10].
PCM can be integrated into PU foam by indirect incorporation of PCM microcapsules into polyurethane mixture at the mixing stage. Microencapsulated paraffin and microencapsulated n-octadecane are the most widely used PCMs loaded into PU foams by indirect method. Incorporation of microencapsulated PCM into PU foam can prevent it from chemical reaction with other ingredients in PU foam and from the possible leakage [1]. However, this method that is extensively used on the laboratory scale increases the manufacturing cost due to need for a pre-synthesis process to encapsulate PCM within a suitable shell material. Furthermore, the shell material results in a decreased specific enthalpy value and reduces the effectiveness of the response to temperature change. The presence of microcapsules also depresses the mechanical properties. The higher microcapsule content, the lower compressive strength the PU foam will have [1, 11]. To cope with the drawbacks, attempts have been made to fabricate PU foam with direct inclusion of PCM material during mixing stage of PU components based on the so-called one-shot synthesis method [6], which allows PCM to be embedded in the porous foam being polymerized through adsorptive capacity and capillary action [1]. Some researchers also employed adsorptive capacity and capillary action to entrap PCM via impregnation of already consolidated PU foams with liquid PCM [6]. This post-synthesis method has been used for loading aqueous saturated inorganic salt hydrate solutions as PCMs. The main drawback of structures produced by impregnation method limiting their application in TES systems is their susceptibility to decomposition after several thermal cycles, phase segregation and supercooling [6]. Various types of polyethylene glycol (PEG) were also loaded into already coalesced PU foam sheets via impregnation method [12]. In general, this method is suitable for loading PCMs such as PEGs whose performance might be impaired by being involved in PU polymerization reaction.
Taking the advantages of cost-effectiveness, simplicity, and enhanced total heat absorption capacity, Sarrie and Onder incorporated n-alkanes, including n-hexadecane and n-octadecane directly into PU foams at different ratios [13]. In the case of one-shot synthesis, there was concern that PCM-PU composite foams may suffer from PCM leakage problem in its liquid state [1, 6]. However, Sarrie and Onder reported no leakage of n-octadecane from PU foams which brought them to this conclusion that the leakage behavior is more likely for PCMs consisting of smaller molecules. In addition, the leakage possibility of PCM material will decrease as the honeycomb of PU foam becomes firmer [13]. Aydin and Okutan also employed the direct method to incorporate a fatty acid-ester-based PCM, i.e. myristyl myristate dissolved in tetrahydrofuran (THF) into PU foam at different ratios [11]. They also showed a negligible leakage due to well encapsulation of PCM in the crosslinked PU matrix. Although both studies in which a direct incorporation method was used for incorporation of PCM into PU foams have reported promising results in terms of thermal properties of PU-PCM composite, there is a lack of information about the effect of PCM materials loaded directly into PU foam on mechanical properties of the foam. In the case of n-octadecane as a PCM directly incorporated into PU foam, thermal behavior during cooling process is also unavailable.
Therefore, in this work, we fabricated PU-PCM composite foams by directly incorporation of n-octadecane in different weight ratios and investigated the effect of PCM content on chemical, morphological, thermal, and mechanical properties of the foams.
Experimental
Materials
Both precursors including a mixture of polyether and brominated polyols, activators containing amine, silicon-based stabilizer, blowing agent and flame retardants (617 FR, viscosity at 25 °C = 2000–2400 mPa.s, density at 25 °C ~ 1.08 g/cm3, OH value: 390–410 mgKOH/g) and methylene diphenyl diisocyanate (MDI, viscosity at 25 °C ~ 160–240 mPa.s, density at 20 °C ~ 1.23 g/cm3, NCO content: 30.5–32.5% by wt.) were provided by Arian Polyurethane JSC (Iran). N-octadecane utilized as PCM was purchased from Merck.
Synthesis of PU-PCM composite foams
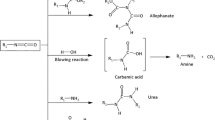
The liquid precursors in a weight ratio of 1:1 was used for synthesis of PU foams. First, the proper amount of melted PCM was mechanically mixed with MDI for 120 s at 1300 rpm. Then, it was added to the mixture containing polyols, activators, stabilizer, blowing agent and flame retardants and vigorously mixed in a beaker using a bench scale overhead stirrer for 40 s at 1300 rpm. The resulting mixture was then poured in a mold and placed at room temperature for 24 h to allow complete foaming and further curing. The synthesis procedure is illustrated schematically in Fig. 1. The pristine PU foam was also synthesize as control. Table 1 summarizes the ingredients and adopted nomenclature of prepared samples.

Schematic illustration of one-shot synthesis of PU-PCM composite foams
Characterization
Olympus SZX16 stereo microscope was used to take images of synthesized foams in order to investigate the effect of PCM molecules on the morphological characteristics of composite foams in comparison with those of pristine PU foam.
FTIR spectra were recorded on Nicolet iS20 FTIR spectrometer (Thermo Fisher Scientific) between the wavenumbers of 4000 and 600 cm−1.
X-ray diffraction (XRD) tests were conducted for n-octadecane, PUF and PUCF15 samples on a PW1730 diffractometer (Philips). The Cu-Kα (λ = 0.154 nm) generator system was operated at 40 kV and 30 mA, and the scanning 2θ ranged from 10 to 80° with a scanning rate of 3°.min−1.
Phase change properties of composite foams including the latent heat and phase transition temperature were studied by differential scanning calorimetry (DSC1, Mettler Toledo) under nitrogen atmosphere within a certain temperature interval ranging from 0 to 60 °C at 2 °C.min−1. Stoichiometric enthalpy value was also calculated for each composite foam based on the measured latent heat of melting of pure n-octadecane. Then, the loading efficiency percentage indicating the amount of n-octadecane that has been successfully loaded into PU foam was calculated using the Eq. (1):
where ∆Hmeasured is the melting enthalpy of PU-PCM composite foam measured using DSC analysis and ∆Hcalculated is the stoichiometric melting enthalpy of PU-PCM composite foam calculated based on the latent heat of melting of n-octadecane.
Thermal reliability was determined by a thermal cycling test. The samples were placed in a closed thermal cycling test chamber (ATM 7004, Arya Sarmayesh, Iran) and 100 melting/crystallizing cycles were conducted within the temperature interval ranging from 5 to 40 °C at 3.5 °C.min−1. The changes in the latent heat and phase change temperature after thermal cycling tests were measured using DSC under the above analysis procedure.
Specific heat capacity of prepared foams was determined by DSC according to ASTM E1269-11 standard test method. The thermal stability of the foams was studied by using a thermal gravimetric analyzer (TGA, BÄHR- STA-503) at a heating rate of 10 °C.min−1 in the range of 25–600 °C under a nitrogen atmosphere.
Thermal conductivity of PUF and PUCF15 samples was measured by a KD2 Pro (Decagon) using the single needle KS-1 probe. Three replicate were carried out for each foam and a time interval of 15 min was used between measurements to allow for thermal gradients to dissipate.
Leakage behavior of PCM in liquid state from the foams were examined gravimetrically by exposure of the composite foams to elevated temperatures. Briefly, a piece of each foam with a specific weight (ms) and a cross-sectional area of 10 cm2 was placed between two pieces of pre-weighed filter papers (m0) (n = 3). The system was placed in an oven at 30 °C for 120 min followed by 60 min at 40 °C and compressed by 12 kPa static load. The filter papers were then removed and weighed again (m1). The leakage percentage was calculated using Eq. (2).
where ω is mass fraction of PCM used in synthesis of PU-PCM composite foams and was considered as 0.1, 0.15, and 0.2 for PUCF10, PUCF15, and PUCF20 samples, respectively.
Uniaxial compressive test was carried out according to ASTM D1621 (STM-20, Santam, Iran) to investigate the effect of PCM on the mechanical properties of composite foams with dimensions of 60 × 60 × 30 mm3 (n = 3). The crosshead speed was 5.0 mm.min−1 and parallel to the foam rising direction. According to ASTM D1621 standard test method, stress value at 10% deformation was considered as the compressive strength. The specific compressive strength and specific compressive modulus of foams were calculated by dividing compressive strength and compressive modulus by their respective apparent density measured according to ASTM D1622.
To investigate the effect of temperature on the compressive behavior of the PU-PCM composite foams compared to that of PUF, dynamic mechanical thermal analysis (DMTA) was conducted (DMA 1, Mettler Toledo) in compression mode at a frequency of 1 Hz and a heating rate of 10 °C.min−1 in the range of 10–70 °C.
Statistical analysis
All data are expressed as mean ± standard deviation (SD). One-way ANOVA followed by Tukey's post hoc test was used for multiple comparison between groups (S Plus 8.0). For all analyses, significance was assigned for P < 0.05.
Results and discussion
Morphological characteristics
As shown in Fig. 2, the inclusion of PCM influenced greatly the morphological characteristics of rigid PU foams. Pristine PU foam possesses a closed-cell structure with near spherical cells. When PCM was added, microstructure was changed to an irregular structure with decreased closed-cell content. Additionally, inclusion of PCM significantly decreased the cell density and increased the cell size. The negative effect on the foaming system as a result of diffusion of n-octadecane out of the nanocapsules into PU matrix during foam synthesis was also reported by Liang et al. [14]. It might be attributed to the fact that on the one hand, heat of the polymerization reaction is absorbed by n-octadecane which increases the viscosity of the mixture. Therefore, the contact between the two reactants becomes difficult and the progress of polymerization reaction is disturbed. As a result, the driving force for cell nucleation in composite foams decreases compared to that of pristine PU foam. The driving force for cell nucleation is a thermodynamic instability caused by a sudden reduction of gas solubility in the mixture as the polymer chain grows [15]. On the other hand, the presence of n-octadecane can increase the flexibility and extensibility of the cell wall. Therefore, the bubble growth can be faster than curing reactions. Consequently, the lower resistant against the cell growth may result in the foam morphology degeneration (larger cell size, cell rupture and decreased closed-cell content) by coalescence and/or Ostwald ripening [16].

Stereo microscopic images of pristine PU and PU-PCM composite foams
However, the cell size of the composite foams tends to decrease with increasing PCM content. In fact, the interface formed between two immiscibile liquids including n-octadecane and polyurethane precursors plays the role of nucleation sites, reduces the nucleation energy and increase the nucleation rate [17]. As the number of nucleation sites increases, the distance among them decreases, leading to the less space for cell growth which results in the reduced cell size [18]. As the PCM content increases, n-octadecane molecules can promote heterogeneously nucleation of the foam cells and cause the structural irregularity and decrease of the cell size. Nevertheless, the cell size of PUCF20 is still significantly larger than that of pristine PU foam.
Chemical structure and crystallization performance
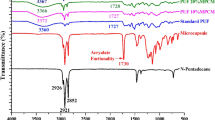
The FTIR spectra of n-octadecane, PU foam and PU-PCM composite foams are shown in Fig. 3a. The FTIR spectra of synthesized foams showed characteristic bands of polyurethane. The absorption bands of hydrogen-bonded N–H with oxygen of ether and oxygen of carbonyl in the urethane which occur at 3310–3290 cm−1 and 3350–3300 cm−1, respectively [19] resulted in a broad peak in this region which can be overlapped with the absorption band associated with -OH stretching vibration of non-bonded polyol at 3600–3100 cm−1. The C = O stretching in carbamate group is detected at 1740–1683 cm−1 [20]. In addition, the bands at 1217 cm−1 and 1062 cm−1 are attributed to the stretching vibrations of C–N and C–O which confirm the formation of urethane linkage between hydroxyl groups of polyol and –NCO [21]. The bands at 1594 cm−1 and 1508 cm−1 are derived from the aromatic rings of MDI [21].

(a) FTIR spectra of pure n-octadecane, pristine PU, and PU-PCM composite foams. (b) Normalized FTIR spectra of pristine PU and PU-PCM composite foams between the wavenumbers of 2350 and 2150 cm−1 by referring to phenyl band at 1594 cm−1. (c) XRD patterns of pure n-octadecane, PUF, and PUCF15
The absorption peaks of n-alkanes occurring around 2953 cm−1, 2914 cm−1, and 2849 cm−1 which are related to the CH3 asymmetrical stretching, methylene asymmetric C–H stretching, and CH2 symmetrical stretching, respectively [22] are detected in FTIR spectra of n-octadecane as well as respective spectra of PU-PCM composite foams. By increasing the PCM content from 10 wt.% to 20 wt.%, the intensity of these peaks increased as well which confirms capturing more n-octadecane molecules within PUCF20 foam followed by PUCF15 compared with PUCF10. In addition, the scissoring mode of the CH2 group gives rise to a characteristic band near 1465 cm−1 in IR spectra which often overlaps with CH3 asymmetrical bending in the 1470–1430 cm−1 region [22]. The visibility of this peak in spectrum of PUCF20 also affirms the higher loading of PCM within PUCF20 sample compared with PUCF10 and PUCF15 samples.
The band around 2270 cm−1 represents the excess of –NCO group of diisocyantes which was used to investigate the effect of PCM on polyurethane polymerization. The intensity of this peak is proportional to the isocyanate groups that have not participated in polymerization reaction and can be used to quantitatively compare the unreacted isocyanate in PU and PU-PCM composite foams. To do this, the FTIR spectra between the wavenumbers of 2350 and 2150 cm−1 were normalized by referring to phenyl band at 1594 cm−1. As shown in Fig. 3b, the presence of PCM molecules resulted in increased unreacted isocyanate groups indicating incompleteness of PU foaming in presence of PCM [13] which was affirmed earlier through the morphological characterization.
The crystallization behaviors of n-octadecane, PUF, and PUCF15 were characterized by XRD as shown in Fig. 3c. It can be seen that PUF has a broad diffraction band at 2θ = 10–30° with a maximum peak appeared at approximately 20°, suggesting the PUF sample is amorphous [23]. For n-octadecane, four characteristic diffraction peaks of triclinic crystal phase appeared at 2θ = 19.5, 20.1, 23.5, and 25.1°, indexed as (011), (011), (100), and (111), respectively [24].
X-ray diffraction patterns observed for PUCF15 present the convolution of the broad PU component peak and the PCM peaks from 10 to 30° which provides evidence that PCM was successfully loaded into PUCF15.
Thermal properties
Thermal properties of n-octadecane including phase change behavior, specific heat capacity changes, and TGA curves are shown in Figs. S1 and S2. The phase change behavior of PU foam, and PU-PCM composite foams was investigated by DSC method, and the obtained DSC curves are shown in Fig. 4a, b.

DSC curves illustrating phase change behavior of pristine PU and PU-PCM composite foams (a) during the first heating scan, (b) during the first cooling scan and (c) during the heating scan after application of 100 thermal cycles. (d) Specific heat capacity changes of pristine PU and PU-PCM composite foams
The phase change peak temperature and phase change enthalpy during the first heating scan as well as loading efficiency are listed in Table 2. Figure 4a shows that there is no phase change for the PUF sample in the temperature range of 0–60 °C. As shown in Fig. S1, n-octadecane has an intensive endothermic melting peak at 29.1 °C. The melting process of n-octadecane in all composite foams took place in one step around 27 °C and the melting enthalpy increased by increasing the PCM content from 17.72 J.g−1 measured for PUCF10 to 24.39 J.g−1 for PUCF15 and to 34.51 J.g−1 for PUCF20 (Fig. 4a and Table 2) which are considerably higher than enthalpy values reported for PU foams containing a similar amount of n-octadecane in the micro/nanoencapsulated form [14, 25,26,27,28,29,30].
Considering a melting enthalpy of 211 J.g−1 for n-octadecane (Fig. S1), the efficiency of PCM loading into PU foams was calculated as 77%-84%. Composite foam containing 30 wt.% PCM was also prepared and its loading efficiency was found as 16%. Therefore, 20 wt.% PCM content was considered as the maximum amount of PCM that was used for synthesis of PU-PCM composite foams.
While, there was one exothermic peak in the cooling curve of n-octadecane at 22.6 °C (Fig. S1) caused by the direct transition from isotropic liquid phase to ordered triclinic phase [31], PU-PCM foams exhibited 2–3 exothermic peaks. The crystallization process of n-alkanes is dominated by the heterogeneous nucleation mechanism. The nucleation proceeds through a transient metastable rotator phase and the stable phase observed for C16, C18, and C20 is triclinic crystalline phase [32]. The heterogeneous nucleation of encapsulated n-octadecance results in two crystallization peaks [32]. These peaks from high to low temperature are called as α and β which are ascribed to heterogeneously nucleated liquid-rotator transition and rotator-triclinic crystal phase transition, respectively. Homogeneously nucleated liquid–crystal phase transition can cause a third crystallizing peak called as γ in cooling curve of n-octadecane [30, 33, 34]. As it can be seen in Fig. 4b, the heterogeneous nucleation peaks are quite variable [35]. The enthalpy sum of the crystallization peaks was obtained as 16.26, 21.85, and 38.09 J.g−1 for PUCF10, PUCF15, and PUCF20, respectively that are comparable to the respective melting enthalpy values. The crystallization temperature of n-octadecane decreased after confinement in porous PU foams which can be attributed to the low thermal conductivity of foam structures. As the foam cell size decreased by increasing the PCM content from 10 wt. % to 20 wt. %, the degree of supercooling defined as the difference between the melting and the crystallization temperatures for different peaks (∆Tα, ∆Tβ, and ∆Tγ) [32] has increased as well. Minimizing the degree of supercooling of n-octadecane incorporated in PU matrix is highly desirable for the energy storage applications [32] which will be addressed in our future study.
To evaluate the thermal reliability of PU-PCM composite foams during repetitious melting/solidifying processes, 100 thermal cycles in the range of 5 to 40 °C were applied. Figure 4c illustrates the DSC curves of PU-PCM composite foams during heating scan after application of thermal cycles. The phase change peak temperatures and phase change enthalpy after 100 thermal cycles are presented in Table 2. It is clear that the melting temperature did not substantially change after the application of thermal cycles. Although, PUCF10 samples could retain 79% of their integrity after the application of cycling thermal loads, the results obtained for PUCF15 and PUCF20 samples were more promising and the measured melting enthalpy decreased by 12% and 16% for PUCF15 and PUCF20, respectively compared to respective melting enthalpy measured during the first heating scan. This result suggests that there is less concern about the PCM leakage from PUCF15 and PUCF20 samples.
In fact, PCMs undergo both sensible and latent heat processes based on the instantaneous PCM temperature compared to the melting/freezing range. The total heat storage capacity of an LHS system is given by Eq. (3) [36]:
where ti is the initial temperature, tm, is the melting temperature and tf is the final temperature all in (°C), m is the mass of PCM medium in (g), Cpi is the average specific heat capacity between Ti and Tm and Cps is the average specific heat capacity between Tm and Tf both in (J.g−1.°C−1); am is the fraction of melted PCM, and ∆Hm is the latent heat of melting in (J.g−1). Therefore, an additional DSC analysis was carried out to evaluate the heat absorption capacity improvement provided by PCM in terms of specific heat. Fig. S1 and Fig. 4d illustrate the specific heat capacity changes of pure n-octadecane, pristine PU foam and PU-PCM composite foams. The specific heat capacity-temperature curve of PUF sample in the range of 10–50 °C is almost a straight line with an average specific heat capacity of 1.40 J.g−1.°C−1. However, the specific heat capacity-temperature curves of pure n-octadecane and PU-PCM composite foams exhibited a maximum value around their melting temperatures as 25.70 J.g−1.°C−1 for pure n-octadecane, 3.28 J.g−1.°C−1 for PUCF10, 4.10 J.g−1.°C−1 for PUCF15, and 4.44 J.g−1.°C−1 for PUCF20.
Thermal stabilities of n-octadecane, PU and PU-PCM composite foams investigated by TGA measurement are shown in Figs. S2 and 5a. As illustrated in Fig. S2, the weight loss for the pure n-octadecane in the temperature range of 135–255 °C is attributed to the evaporation of n-octadecane. The pristine PU foam undergoes a one-step thermal degradation while PU-PCM composite foams exhibit clearly two degradation steps corresponding to the evaporation of the PCM and degradation of the PU matrix (Fig. 5a).

(a) TGA curves of pristine PU and PU-PCM composite foams. (b) Leakage test results for PU-PCM composite foams (* indicates significant differences between PUCF10 and PUCF20 at P < 0.05)
The temperature associated to a weight loss of 5% (T0.05) was considered as the onset temperature of weight loss of PCM [37, 38]. For PU-PCM composite foams, T0.05 is influenced by two effects. On the one hand, the evaporation of n-octadecane can be delayed due to the low thermal conductivity of PU matrix and protection provided by PU matrix. On the other hand, n-octadecane molecules are distributed over the PU matrix in smaller aggregates with higher surface area compared to pure n-octadecane aggregates which can consequently lead to the reduced evaporation temperature of n-octadecane. As a consequence of these conflicting effects, the onset temperature of weight loss increased from 195 °C for pure n-octadecane to 221 °C and 210 °C for PUCF10 and PUCF15, respectively, while it decreased to 190 °C for PUCF20 (Table 3).
The pristine PU foam exhibits the most durable behaviour among the samples. Considering the mass residue at 600 °C, it can be found that the residue of the PUF is about 29.3% and the pyrolyzed ash content decreases with increasing the PCM content (Table 3). Aydin et al. also have reported similar results [11]. It might be due to the higher crosslinking density of PUF sample affirmed by the normalized FTIR spectra (Fig. 3b) facilitating formation of a carbonaceous char on the surface of the sample which can impede further degradation of the sample core at elevated temperatures [37, 39]. In fact, the decomposition of urethane bonds results in the formation of alcohol and isocyanate groups. Dimerization of isocyanate produces carbodiimides which react with free alcohol groups and leads to the formation of thermally stable substituted urea that is converted to the carbonaceous char in the last step of degradation process [40].
PU foam has a thermal conductivity of 0.023 W.m−1.K−1. Addition of PCM to PU foam could significantly improve the thermal conductivity, so that PUCF15 showed a considerable higher conductivity as 0.045 ± 0.005 W.m−1.K−1 (P < 0.05).
Leakage behavior
Leakage behavior of PU-PCM composite foams is a critical property affecting production, handling and applications [13]. No oil spots were observed on the filter paper sheets after the leakage test. In addition, Fig. 5b shows the results of leakage test conducted for composite foams. As it can be seen the PCM leakage from PUCF10 is 9.0 ± 1.7% which is a worrying value. As the PCM content increased, and the foam structure became firmer, the leakage decreased and reached 6.3 ± 0.7% and 5.9 ± 0.8% from PUCF15 and PUCF20 samples, respectively which shows significant reduction in leakage from PUCF20 samples compared to that from PUCF10 samples (P < 0.05). This result is in agreement with results obtained after thermal cycling and indicates that directly employed PCM can properly encapsulate in the matrix during formation of PUCF20 samples.
Compressive properties
The mechanical properties of polyurethane foams are affected by foam density and geometry of foam cells [41]. As approved by morphological analysis and FTIR and as reported by others [13], directly incorporation of PCM interfered with the foaming reactions which resulted in increased density of PU-PCM composite foams compared to that of pristine PU foam. Therefore, to eliminate the effect of variation in density, compressive properties of each foam were normalized by its corresponding density. The specific compressive properties of prepared foams including specific compressive strength (S10%) and specific compressive modulus are shown in Fig. 6a. Both specific compressive strength and specific compressive modulus values of PU-PCM composite foams are lower than those of pristine PU foam (3.89 ± 0.20 kPa.m3.kg−1 and 77.48 ± 5.72 kPa.m3.kg−1, respectively) which can be due the reduced closed cell content in PU-PCM composite foams. After incorporation of 10 wt.% PCM, specific compressive strength and modulus of PU foam dropped to 2.67 ± 0.43 kPa.m3.kg−1 and 46.05 ± 9.86 kPa.m3.kg−1, respectively. The defects on the closed-cell impair the mechanical performance of polyurethane foams [42]. On the other hand, as the PCM content increased, the specific compressive strength and modulus increased and reached 3.44 ± 0.30 kPa.m3.kg−1 and 65.50 ± 1.16 kPa.m3.kg−1, respectively for PUCF20 which are comparable to specific compressive properties obtained for PUF. These improvements can be attributed to the formation of smaller cell size by increasing the PCM content which was confirmed by morphological analysis. Foams with smaller cell size and more cells can withstand higher external loads due to more struts per unit area of foam to support structure under loading [42, 43]. Since incorporation of micro/nanoencapsulated PCM decreases the mechanical strength compared with that of pristine PU foams [1], this promising result shows superior capability of one-shot synthesized PU-PCM composite foams provided that appropriate amount of PCM is incorporated.

(a) Specific compressive strength and specific compressive modulus of pristine PU and PU-PCM composite foams (* and + indicate significant differences in specific compressive strength and specific compressive modulus, respectively compared to those of PUF at P < 0.05). (b) Storage modulus (E′) of pristine PU and PU-PCM composite foams as a function of temperature obtained by DMTA in compression mode
The mechanical tests were carried out at room temperature when the PCM incorporated into foams was in solid state. It is worth mentioning that at temperatures higher than melting temperature of PCM when it is mostly in liquid phase, the compressive properties of PU-PCM composite foams might be further reduced because melted PCM can serve as the plasticizer. To investigate this issue, the evolution of the storage modulus (E′) as a function of temperature has been measured by DMTA in compression mode (Fig. 6b). As expected, a decrease of E′ is observed for PU-PCM composite foams with the increase of the temperature. With increasing the PCM content from 10 wt.% to 20 wt.%, the E′ reduction increases accordingly. Another important result is that the storage modulus (E′) of PUF and PU-PCM composite foams show the same trend as the specific compressive modulus obtained from the uniaxial compressive test, leading to the same comment on the effect of PCM content.
Conclusion
Thermoregulated rigid PU foams were synthesized by directly incorporation of PCM into PU matrix. The successful incorporation of PCM was confirmed by FTIR spectra, XRD, and DSC analysis. FTIR analysis also demonstrated that inclusion of PCM negatively effects the foaming process. Correspondingly, the foam density increased by increasing the PCM content. Moreover, as the PCM content increased, thermal energy storage capacity of composite foams was enhanced. The phase change enthalpies of PU-PCM composite foams ranged from 17.72- 34.51 J.g−1 corresponding to the loading efficiencies of 77- 84%. The composite foams also possessed good thermal reliability after 100 thermal cycles. The presence of PCM in composite foams had a major impact on cell structure which consequently affected the mechanical properties. Although, inclusion of PCM increased significantly the cell size and reduced the closed-cell content of the composite foams compared to those of the pristine PU foam, a decreasing trend was observed in the cell size of composite foams in response to increasing the PCM content. As a consequence, composite foams showed reduced specific compressive strength and specific compressive modulus compared to those of pristine PU foam, though, as the PCM content increased, compressive properties were improved due to the reduced cell size. In addition, as the PCM content increased, and the foam structure became firmer, the leakage decreased and there was less concern about the PCM leakage.
Therefore, one-shot synthesized PU-PCM composite foams can take advantages of cost-effectiveness, improved thermal storage capacity, sufficient mechanical properties, and reduced leakage provided that appropriate amount of PCM (20 wt.% in this study) is incorporated. However, they may suffer from the problem of supercooling which needs to be addressed in future works.
References
Yang C, Fischer L, Maranda S, Worlitschek J (2015) Rigid polyurethane foams incorporated with phase change materials: A state-of-the-art review and future research pathways. Energy Build 87:25–36. https://doi.org/10.1016/j.enbuild.2014.10.075
Baetens R, Jelle BP, Gustavsen A (2010) Phase change materials for building applications: A state-of-the-art review. Energy Build 42:1361–1368. https://doi.org/10.1016/j.enbuild.2010.03.026
Kalnæs SE, Jelle BP (2015) Phase change materials and products for building applications: A state-of-the-art review and future research opportunities. Energy Build 94:150–176. https://doi.org/10.1016/j.enbuild.2015.02.023
Marin P, Saffari M, de Gracia A et al (2016) Energy savings due to the use of PCM for relocatable lightweight buildings passive heating and cooling in different weather conditions. Energy Build 129:274–283. https://doi.org/10.1016/j.enbuild.2016.08.007
Saffari M, De Gracia A, Ushak S, Cabeza LF (2016) Economic impact of integrating PCM as passive system in buildings using Fanger comfort model. Energy Build 112:159–172. https://doi.org/10.1016/j.enbuild.2015.12.006
Ikutegbe CA, Farid MM (2020) Application of phase change material foam composites in the built environment: A critical review. Renew Sustain Energy Rev 131:110008. https://doi.org/10.1016/j.rser.2020.110008
Ahmadi Y, Kim K-H, Kim S, Tabatabaei M (2020) Recent advances in polyurethanes as efficient media for thermal energy storage. Energy Storage Mater 30:74–86. https://doi.org/10.1016/j.ensm.2020.05.003
Amaral C, Pinto SC, Silva T et al (2020) Development of polyurethane foam incorporating phase change material for thermal energy storage. J Energy Storage 28:101177. https://doi.org/10.1016/j.est.2019.101177
Delgado JMPQ, Martinho JC, Vaz Sá A, et al (2019) Thermal Energy Storage with Phase Change Materials, A Literature Review of Applications for Buildings Materials. 73
Liao H, Liu Y, Chen R, Wang Q (2021) Preparation and characterization of polyurethane foams containing microencapsulated phase change materials for thermal energy storage and thermal regulation. Polym Int 70:619–627. https://doi.org/10.1002/pi.6145
Aydin AA, Okutan H (2013) Polyurethane rigid foam composites incorporated with fatty acid ester-based phase change material. Energy Convers Manag 68:74–81. https://doi.org/10.1016/j.enconman.2012.12.015
Sarier N, Onder E (2008) Thermal insulation capability of PEG-containing polyurethane foams. Thermochim Acta 475:15–21. https://doi.org/10.1016/j.tca.2008.06.006
Sarier N, Onder E (2007) Thermal characteristics of polyurethane foams incorporated with phase change materials. Thermochim Acta 454:90–98. https://doi.org/10.1016/j.tca.2006.12.024
Liang S, Zhu Y, Wang H et al (2016) Preparation and Characterization of Thermoregulated Rigid Polyurethane Foams Containing Nanoencapsulated Phase Change Materials. Ind Eng Chem Res 55:2721–2730. https://doi.org/10.1021/acs.iecr.5b04543
Minogue E (2000) An in-situ study of the nucleation process of polyurethane rigid foam formation. Dublin City University
Brondi C, Di Maio E, Bertucelli L et al (2021) Competing bubble formation mechanisms in rigid polyurethane foaming. Polymer (Guildf) 228:123877. https://doi.org/10.1016/j.polymer.2021.123877
Vehkamäki, H (2006) Classical Nucleation Theory in Multicomponent Systems. Springer-Verlag, Berlin/Heidelberg
Sánchez-Calderón I, Bernardo V, Santiago-Calvo M et al (2021) Effect of the Molecular Structure of TPU on the Cellular Structure of Nanocellular Polymers Based on PMMA/TPU Blends. Polymers (Basel) 13:3055. https://doi.org/10.3390/polym13183055
Wong CS, Badri KH (2012) Chemical Analyses of Palm Kernel Oil-Based Polyurethane Prepolymer. Mater Sci Appl 03:78–86. https://doi.org/10.4236/msa.2012.32012
Lin-Vien D, Colthup NB, Fateley WG, Grasselli JG (1991) CHAPTER 9 - Compounds Containing the Carbonyl Group. In: Lin-Vien D, Colthup NB, Fateley WG, Grasselli JG (eds) The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules. Academic Press, San Diego, pp 117–154
Li S, Li C, Li C et al (2013) Fabrication of nano-crystalline cellulose with phosphoric acid and its full application in a modified polyurethane foam. Polym Degrad Stab 98:1940–1944. https://doi.org/10.1016/j.polymdegradstab.2013.06.017
Lin-Vien D, Colthup NB, Fateley WG, Grasselli JG (1991) CHAPTER 2 - Alkanes. In: Lin-Vien D, Colthup NB, Fateley WG, Grasselli JG (eds) The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules. Academic Press, San Diego, pp 9–28
Pinto ERP, Barud HS, Polito WL et al (2013) Preparation and characterization of the bacterial cellulose/polyurethane nanocomposites. J Therm Anal Calorim 114:549–555. https://doi.org/10.1007/s10973-013-3001-y
Xie B, Liu G, Jiang S et al (2008) Crystallization Behaviors of n -Octadecane in Confined Space: Crossover of Rotator Phase from Transient to Metastable Induced by Surface Freezing. J Phys Chem B 112:13310–13315. https://doi.org/10.1021/jp712160k
You M, Zhang XX, Li W, Wang XC (2008) Effects of MicroPCMs on the fabrication of MicroPCMs/polyurethane composite foams. Thermochim Acta 472:20–24. https://doi.org/10.1016/j.tca.2008.03.006
You M, Zhang X, Wang J, Wang X (2009) Polyurethane foam containing microencapsulated phase-change materials with styrene-divinybenzene co-polymer shells. J Mater Sci 44:3141–3147. https://doi.org/10.1007/s10853-009-3418-7
You M, Zhang XX, Wang XC et al (2010) Effects of type and contents of microencapsuled n-alkanes on properties of soft polyurethane foams. Thermochim Acta 500:69–75. https://doi.org/10.1016/j.tca.2009.12.013
Tinti A, Tarzia A, Passaro A, Angiuli R (2014) Thermographic analysis of polyurethane foams integrated with phase change materials designed for dynamic thermal insulation in refrigerated transport. Appl Therm Eng 70:201–210. https://doi.org/10.1016/j.applthermaleng.2014.05.003
Qu L, Li A, Gu J, Zhang C (2018) Thermal Energy Storage Capability of Polyurethane Foams Incorporated with Microencapsulated Phase Change Material. Chemistry Select 3:3180–3186. https://doi.org/10.1002/slct.201703043
Zhu Y, Qin Y, Liang S et al (2019) Nanoencapsulated phase change material with polydopamine-SiO 2 hybrid shell for tough thermo-regulating rigid polyurethane foam. Thermochim Acta 676:104–114. https://doi.org/10.1016/j.tca.2019.04.005
Cholakova D, Denkov N (2019) Rotator phases in alkane systems: In bulk, surface layers and micro/nano-confinements. Adv Colloid Interface Sci 269:7–42. https://doi.org/10.1016/j.cis.2019.04.001
Zhang XX, Fan YF, Tao XM, Yick KL (2005) Crystallization and prevention of supercooling of microencapsulated n-alkanes. J Colloid Interface Sci 281:299–306. https://doi.org/10.1016/j.jcis.2004.08.046
Wang X, Li C, Wang M et al (2020) Bifunctional microcapsules with n-octadecane/thyme oil core and polyurea shell for high-efficiency thermal energy storage and antibiosis. Polymers (Basel) 12:1–16. https://doi.org/10.3390/polym12102226
Woo HY, Lee DW, Yoon TY et al (2021) Sub-100-nm nearly monodisperse n-paraffin/pmma phase change nanobeads. Nanomaterials 11:1–9. https://doi.org/10.3390/nano11010204
Oliver MJ, Calvert PD (1975) Homogeneous nucleation of n-alkanes measured by differential scanning calorimetry. J Cryst Growth 30:343–351. https://doi.org/10.1016/0022-0248(75)90010-X
Faraj K, Khaled M, Faraj J et al (2020) Phase change material thermal energy storage systems for cooling applications in buildings: A review. Renew Sustain Energy Rev 119:109579. https://doi.org/10.1016/j.rser.2019.109579
Galvagnini F, Dorigato A, Valentini F, Fiore V, La Gennusa M, Pegoretti A (2020) Multifunctional polyurethane foams with thermal energy storage/release capability. J Therm Anal Calorim 1–17. https://doi.org/10.1007/s10973-020-10367-w
Qiu X, Lu L, Tang G, Song G (2021) Preparation and thermal properties of microencapsulated paraffin with polyurea/acrylic resin hybrid shells as phase change energy storage materials. J Therm Anal Calorim 1–10. https://doi.org/10.1007/s10973-020-09354-y
Liu X, Hao J, Gaan S (2016) Recent studies on the decomposition and strategies of smoke and toxicity suppression for polyurethane based materials. RSC Adv 6:74742–74756. https://doi.org/10.1039/C6RA14345H
Alobad ZK, Albozahid M, Naji HZ et al (2021) Influence of hard segments content on thermal, morphological and mechanical properties of homo and co-polyurethanes: a comparative study. Arch Mater Sci Eng 1:5–16. https://doi.org/10.5604/01.3001.0015.0510
Silva MC, Takahashi JA, Chaussy D et al (2010) Composites of rigid polyurethane foam and cellulose fiber residue. J Appl Polym Sci 117:3665–3672. https://doi.org/10.1002/app.32281
Leng W, Pan B (2019) Thermal insulating and mechanical properties of cellulose nanofibrils modified polyurethane foam composite as structural insulated material. Forests 10:200. https://doi.org/10.3390/f10020200
Fan H, Tekeei A, Suppes GJ, Hsieh FH (2012) Properties of biobased rigid polyurethane foams reinforced with fillers: Microspheres and nanoclay Int J Polym Sci. https://doi.org/10.1155/2012/474803
Acknowledgements
This work was supported by Hormozgan Electric Distribution Company (HEDC) (Project No. 980211) and Shahid Beheshti University. The authors gratefully acknowledge research Laboratories of Shahid Beheshti University, Zirab Campus for technical support and Arian Polyurethane JSC for kindly providing ingredients used for synthesis of polyurethane.
Funding
This work was supported by Hormozgan Electric Distribution Company (HEDC) (Project No. 980211) and Shahid Beheshti University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no known competing financial interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Vatankhah, E., Abasnezhad, M., Nazerian, M. et al. Thermal energy storage and mechanical performance of composites of rigid polyurethane foam and phase change material prepared by one-shot synthesis method. J Polym Res 29, 81 (2022). https://doi.org/10.1007/s10965-022-02911-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-022-02911-z