
Abstract
Bio-composites are known to have poor properties. The reason is the absence of chemical bonding between bio-fillers and polymers. In this present study, we prepared composites with a bio-filler by melt blending distillers grains (DG) with high-density polyethylene (HDPE) and modified HDPE (MHDPE), and compared the characteristics of the composites. Differential scanning calorimetry, thermogravimetry, mechanical property measurement, Fourier transform infrared spectroscopy, contact angle measuring instrument, and water absorption analysis were used to assess DG as a bio-filler. From the results of hydrophilicity measurement, water absorption, morphology, and tensile strength tests, MHDPE was found to have a better interfacial adhesion with DG. The yield strength and crystallinity of DG/MHDPE bio-composites with 50% DG were higher than those of pure HDPE. As DG improved the bio-composite performance, it would have broad application prospects as a bio-filler.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Distillers grains (DG) are a by-product of breweries. They are generally considered an industrial waste [1]. According to the Renewable Fuels Association, the production of ethanol in the USA has increased to 30 billion liters from 1980 to 2010, and DG as a by-product has amounted to 250 million metric tons [2]. In China, the amount of ethanol production increases yearly [3,4,5], which is associated with 25 million tons of DG. Balat et al. [6] indicated that the global production of bio-ethanol in 2000 increased from 170 to 460 l. Proskurina et al. [7] reported in 2019 that the development of bioenergy (from ethanol as biofuel) showed a steady growth trend. With the production of wine and biofuel, there is also a huge quantity of DG being produced, and this amount translates to a lot of waste. Hence, how to dispose of DG efficiently should be a top priority.
Traditional treatments for DG include composting and animal husbandry [8,9,10,11,12]. Leonardi et al. [13] reported that DG as a feedstock reduced the butterfat content of cow. Wu et al. [14] found that using DG as animal feeds was not good because animals may be fed with mycotoxins. Therefore, the traditional ways are not satisfactory enough for the efficient disposal of DG. We need other methods to deal with DG whose accumulation is constantly increasing.
Problems with environmental pollution have been aggravated because so much bio-based fillers are being generated: wood flour [15,16,17,18], DG [19], shell powder [20, 21], bagasse [22, 23], rice husk [24,25,26], eggshell [27], and tapioca [28]. Bio-fillers are renewable, resource-rich, cheap, and eco-friendly, in contrast to inorganic fillers that are non-renewable.
Toro et al. [29] contrasted the different types of fillers (inorganic fillers and bio-fillers), which were incorporated into eggshell/polypropylene composites through melt-blending. The mechanical properties of the resultant composites were more robust because of the reinforcement provided by the eggshell. Tsou et al. [30] used a twin-screw extruder to mix polylactide with rice husk, and added methylene diphenyl diisocyanate as an interfacial compatibilizer; the result was a renewable composite with enhanced tensile strength and biodegradation for 3D printing applications. Hence, biowaste can give improved characteristics to polymer matrices and boost the composite properties.
Polyethylene (PE) is one of most common polymers in the world and has been applied to many fields (e.g., in agriculture, packaging) [31,32,33]. PE was sold for $1.85–2.27/kg (0.91–1.12/lb) in 2012 [34]. If we added DG to PE to form semi-degradable DG/PE composites with a required total weight, the amount of PE that was otherwise required would be reduced, as DG was the other component that would contribute for the total weight; thus, it would lead to cost efficiency.
Studies on DG and polymer composites are few. Even fewer studies on DG/PE composites have been reported (i.e., the use of recycled DG in PE has not been investigated adequately). Luo et al. [35] examined DG as fillers for low density polyethylene (LDPE). They introduced 10–40% DG treated with 5% NaOH solution into the LDPE matrix, and found that DG had great potential application in thermoplastic composites. However, because LDPE lacked functional groups and had poor compatibility with DG, the mechanical properties (elongation at break and tensile strength) of the composites were considerably lower than those of pure HDPE.
To explore the potential application of DG as fillers in HDPE composites and to improve the properties of the composites, we modified HDPE (to give MHDPE) and added a content of DG as high as 50% to the MHDPE matrix. DG from waste disposal sites was recycled and blended with HDPE or MHDPE using a mixer to obtain DG/HDPE or DG/MHDPE composites. The mechanical property, thermostability, crystallinity, and compatibility were determined.
Experiment
Materials
HDPE (LH503) was produced from USI Co., Ltd. (Taipei, Taiwan), whereas HDPE-COOH is similar to that prepared by previous studies [36]. DG came from Sichuan Brewery (China).
Pretreatment of DG
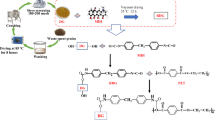
Figure 1 shows the flow for preparing DG/HDPE and DG/MHDPE composites. DG was washed and dried at 80 °C. Afterward, it was ground to a powder form (200–300 mesh) using a vibration mill (Shanghai, China). After several times of crushing and grinding, DG flour (200–300 mesh) was produced. Finally, the DG flour was dried at 105 °C and kept for later use.

Preparation of DG/HDPE and DG/MHDPE composites
Preparation of DG/HDPE and DG/MHDPE composites
Raw materials in varying proportions were weighed and combined to prepare DG-modified composites of HDPE and MHDPE. Table 1 shows the obtained composition. DG/HDPE and DG/MHDPE composites were prepared using a HAAKE mixer (Thermo Fisher Scientific, Germany), which was operated at a speed of 90 rpm and at 160 °C. The composites came out brown in color, and they were vacuum-dried at 80 °C for 8 h, then at 120 °C for 3 h prior to hot-pressing. Thermocompression was implemented at 160 °C and 15 MPa for 3 min. Afterward, the composites were cooled at room temperature. Ultimately, sheets of DG/HDPE and DG/MHDPE were produced.
Characterization
Mechanical tests were performed using a microcomputer-controlled electronic universal testing machine (Furbs, Xiamen Furbusi Testing Equipment Co., Ltd., Xiamen, China), according to the standard method of ASTM D638 Type IV. The test speed was 50 min/mm. Five samples were measured, and the results averaged. The tensile strength (σ, MPa), yield strength (σ, MPa), and elongation at break (ɛ, %) were calculated using the following equations:
where P was the maximum load (MPa); b and d were width and thickness (mm), respectively; G0 was the original gauge length of samples (mm); G was the pitch spacing (mm) when the sample broke.
Differential scanning calorimetry (DSC) of samples was conducted with a DSC-200 F3 calorimeter (Netzsch, Germany) under nitrogen. Samples were sealed hermetically in DSC pans and heated to 300 °C from room temperature at a rate of 10 min/°C, after which they were cooled to room temperature at a speed of 10 °C/min, then reheated to 300 °C with the same heating rate, to examine their melting and crystallization behavior, such as melting temperature (Tm) and crystallization temperature (Tc). The crystallinity (Xc) was evaluated from the following relationship:
where α was the content of DG, ΔHm was the measured melting enthalpy, and ΔHm0 was the enthalpy of 100% HDPE crystals (293 J/g) [37].
Thermogravimetric analysis (TGA) was conducted using TGA instruments (Model STA 409PC, Netzsch, Germany); the heating was done under nitrogen environment from room temperature to 650 °C at a scan rate of 10 °C/min. The thermal decomposition temperature (Td) and DTG (derivative of TGA) were obtained.
The crystalline structure and crystallinity were evaluated using X-ray diffraction (XRD, Bruker-D2 HASER, Germany) equipped with Cu-Kα radiation; the data were recorded every 0.02 s at room temperature, and the test range was 5–90°.
Scanning electron microscopy (SEM, TESCAN, Czech) was used to observe the morphology of the fractured surface of samples subjected to tensile testing. Sample fragments were attached to a sample holder by using conductive adhesives, and were sputtered with a thin layer of gold, which was necessary to obtain distinct surface SEM images.
Fourier-transmission infrared spectroscopy (Nicolet 6700 FTIR spectrometer) was operated in transmission mode over a 4000–400 cm−1 wavelength range, with a resolution of 1 cm−1. Samples of pure HDPE and its composites, which were reduced in powder form, were mixed with KBr and subsequently pressed to shapes of disk.
Contact angles were measured using a contact angle meter (JC2000D, Shanghai, China). Samples were dried in an oven at 80 °C for 8 h before they were mounted on a platform, onto which 2 μL of DI water was dropped from a syringe. Contact angle data were recorded by a computer software.
Tsou et al. [30] described a method of water absorption measurement. All samples were dried in an oven at 80 °C for 8 h, then weighed before they were immersed in DI water for 24 h. After that period, the sample surface was gently dabbed with a tissue to remove the water droplets on the surface. Water absorption was calculated from the following equation:
where W0 was the initial weight and W was the measured weight.
Results and discussion
Mechanical property
Figure 2 presents data on yield strength, tensile strength, and elongation at break of HDPE, MHDPE, and DG/HDPE and DG/MHDPE composites. The tensile properties of MHDPE were shown to be lower than those of the neat HDPE; this result is similar to that of previous studies [36, 38]. After the polymer modification, the mechanical properties were slightly reduced. When MHDPE was blended with DG, the resultant composite exhibited higher yield strength than pure HDPE (Fig. 2a); it means that DG acted as a reinforcement agent in the HDPE matrix, because it increased the ability of the material to recover from permanent deformation. However, both the tensile strength (Fig. 2b) and elongation at break (Fig. 2c) showed a downward trend with increasing amounts of DG. Modified composites had better strength and toughness than pure HDPE. The strength and toughness of the composites decreased because of the weak interfacial binding force between DG as a biomaterial and the HDPE matrix [39, 40]. But the functional group in MHDPE improved the interfacial compatibility between the filler and the host polymer; therefore, DG/MHDPE composites had better mechanical properties than DG/HDPE composites.

Yield strength, tensile strength, and elongation at break of HDPE, MHDPE, and their composites
Differential scanning calorimetry
Figure 3 depicts the second heating and cooling DSC curves. The melting temperatures of the composites were lower than those of the original HDPE (Table 2); this may be due to the addition of DG, which made the crystal size of HDPE and MHDPE smaller [34]. Therefore, when the composites were heated, the crystal was disintegrated early, which led to the decrease in melting temperature.

Differential scanning calorimetry for HDPE and its composites: a second heating curves; b second cooling curves
Table 2 lists the DSC data on initial crystallization temperature (Tc), melting temperatures (Tm1 and Tm2), and peak values in cooling curves (Tp1 and Tp2) for HDPE and its composites. Tm1 and Tc1 (Fig. S1a) and Tp1 (Fig. S1b) were obtained from the first heating and cooling scans, respectively. Tc of composites was higher than that of HDPE. The value of (Tp1-Tc1) for HDPE dropped to 5.2 °C from 10.5 °C, and that of (Tp2-Tc2) to 5.1 °C from 9.5 °C. This drop indicated that DG acted as a nucleating agent, promoting the grain size of HDPE to refine and accelerate the crystallization rate and shorten the cycle of product molding [41,42,43]. On the other hand, the crystallinity of 50(DG)/MHDPE was considerably higher than that of HDPE or the other composites. The role of DG as a nucleating agent, as well as the interfacial adhesion between the bio-filler and the matrix, promoted a higher crystal density of the polymer. The good compatibility between DG and MHDPE promoted even dispersion of DG in MHDPE, and this, in turn, effectively increased the amount of nucleation at the initial stage; thus, the crystallization rate of the polymer was enhanced, and the crystal growth was facilitated. As a result, the crystallinity of DG/MHDPE was relatively high [44].
Thermogravimetric analysis
TGA and DTG curves for HDPE and its composites are represented in Fig. 4, and the data are summarized in Table 3. Because DG was added to the host polymer, the initial degradation decreased. According to Table 3, the initial degradation temperature of pure HDPE was 469.1 °C, but the lowest initial degradation temperature of the composites was 261.9 °C. The reason is because hemicellulose in DG was degraded first, leading to weight loss. DG contained a lot of cellulose and free water; hence, the DTG peak at 141.9–144.0 °C and 351 °C referred to water loss and cellulose degradation, respectively [45]. When the temperature reached 478.7–486 °C, DG, HDPE, and MHDPE began to degrade, causing maximal thermal degradation.

Thermogravimetric curves for HDPE and its nanocomposites: a thermogravimetric analysis; b derivative thermogravimetry
Table 3 also indicates the temperatures at 10 wt% weight loss for HDPE and its composites. These results showed that as the DG was added, the thermal stability of the composites decreased with the increase in DG. Lomakin et al. [46] indicated that the thermal stability of LDPE/cellulose composites was lower than that of the pure sample. So, it is a normal phenomenon for the thermal stability of polymer composites with DG to be lower than that of the neat polymer matrix. In addition, the thermal stability of DG/MHDPE was higher than that of DG/HDPE composites. As reported previously, the adhesion between fillers and polymers had influence on the decomposing behavior of composites [20]. It might be attributed to the stronger adhesion and higher compatibility between DG and MHDPE, which led to better dispersion, and, in turn, better synergy between the two materials. In conclusion based on TGA, the thermal stability of all samples resulted in descending order: HDPE >25(DG)/MHDPE >50(DG)/MHDPE >25(DG)/HDPE >50(DG)/HDPE.
Although the results from TGA indicated that the thermal degradation temperature of composites was lower than that of the pure HDPE, the extreme temperature of HDPE was about 100 °C [47], and the initial cracking temperature in the first stage of all composites in this study was about 260 °C. Therefore, compared with the application of HDPE, the application of composite materials would not be too limited.
X-ray diffraction
Figure 5 shows the X-ray diffraction (XRD) patterns of HDPE and its composites filled with DG. The neat HDPE exhibited obvious reflection peaks at 21.3 and 23.7°, which corresponded to the typical orthorhombic unit cell structures for (110) and (200) reflection planes, respectively. These 2θ values agree with the reported values for HDPE [48, 49]. The two weak peaks at 2θ values of about 29.9 and 36.2° were ascribed to reflection planes (210) and (020), respectively [50]. The two crystalline characteristic peaks for (110) and (200) stayed the same after the addition of DG, demonstrating that the blending with DG did not influence the original crystal structure of the HDPE substrate.

XRD plots of HDPE and its composites
The crystallinity data were 73, 48.8, 53.8, 50.7, and 76.7% (Table 4), which were calculated using a computer software; the trend was the same as that of the DSC data in terms of crystallinity. From Fig. 5, the intensity of 50(DG)/MHDPE was the strongest; DG improved the crystallinity of the polymer. The results showed that DG dispersed much better in MHDPE than in HDPE; more DG contributed to more efficient nucleation, which was beneficial for increasing the crystal density [44]. The DSC results also manifested that DG was a nucleating agent. Therefore, the crystallinity of DG/MHDPE composite was higher than that of HDPE or the other composites.
Scanning electron microscopy
SEM images of fractured surfaces of samples are shown in Fig. 6. The DG particles were not uniformly spread throughout the HDPE matrix, and were separated from the matrix (Fig. 6a, b) because the polar groups in DG repelled the nonpolar groups in HDPE [51, 52]; some defects were observed between DG and HDPE. For the DG/MHDPE composites, the DG particles were partially covered by MHDPE, which indicated that the interface bonding in DG/MHDPE composites was better because MHDPE contained partial polar groups. This morphology results are also similar to the results of previous research on rice husks [26, 30, 37], wood floor [18], tapioca [28], and oyster shell [20], which were blended with the polymer. The cross-section of DG/HDPE composites appeared relatively rough compared with that of the modified DG/MHDPE composites. This result confirmed that concentrations greater than 50 wt% DG in the MHDPE matrix led to higher yield strength, crystallinity, and thermal degradation temperature.

SEM images of DG/HDPE and DG/MHDPE composites: a 25(DG)/HDPE; b 50(DG)/HDPE; c 25(DG)/MHDPE; d 50(DG)/MHDPE
Fourier-transform infrared spectroscopy
Figure 7 illustrates the FTIR spectra of HDPE, DG/HDPE, and DG/MHDPE. HDPE and MHDPE blended with DG exhibited strong peaks in the spectra at absorption bands of 3400 and 1050 cm−1, corresponding to –OH of free water and C–O groups [53, 54], respectively, because there were a lot of hydroxyl groups in DG. DG consists of multiple grains made up of many substances. Its main components are cellulose, hemicellulose, and lignin, so the main functional groups are derived from the –OH groups in cellulose and lignin. The increase in peak intensity at about 3400 cm−1 [55] when DG was added referred to the hydroxyl bond vibration (–OH), arising from the substantial contributions of cellulose in DG. The absorption bands at 1700–1850 cm−1 referred to C=O [56]. A new weak peak at about 1505–1510 cm−1 appeared, referring to C=C of the aromatic skeletal (lignin) resulting from the addition of DG to HDPE [57]. This new peak became strong and shifted to about 1513 cm −1 after MHDPE was blended with DG (Fig. 7b), which might correspond to asymmetric COO− stretching from MHDPE [58, 59]; it appeared when DG was mixed with MHDPE [60]. Therefore, an ester bond formed from the reaction between the carboxyl group in MHDPE and the OH group in lignin or the cellulose in DG. The aforementioned test results proved that DG and MHDPE had a better interfacial adhesion than DG and HDPE.

a FTIR spectra of HDPE, DG/HDPE, and DG/MHDPE composites; b FTIR spectra at 1400–1600 cm−1 range
Contact angle
Figure 8 shows the results of contact angle tests on HDPE, DG/HDPE, and DG/MHDPE. It demonstrated that the values of contact angle for HDPE and MHDPE were similar; the value for MHDPE was slightly lower than that for HDPE. It could be attributed to the functional group from MHDPE, and it could be expected that COOH can enhance the hydrophilicity of the polymers [38, 56, 61]. The contact angle for pure HDPE was the largest. DG contained a large amount of –OH, which made the sample hydrophilic, so that the contact angle as a whole tended to decrease when DG was added. However, the contact angle for MHDPE was higher than that for the unmodified one, because the esterification reaction consumed the partial hydroxyl groups in DG and the carboxyl groups in MHDPE. The formation of ester bonds was confirmed by the FTIR results.

Contact angle data: (a) HDPE; (b) MHDPE; (c) 25(DG)/HDPE; (d) 25(DG)/MHDPE; (e) 50(DG)/HDPE; (f) 50(DG)/MHDPE
Water absorption
Figure 9 displays the water absorption for HDPE and for DG/HDPE and DG/MHDPE composites. It indicates that the water absorption data for HDPE and MHDPE were almost the same. It also indicates a trend of increasing water absorption with the DG content. DG made the water absorption rate higher because DG as a bio-filler contained a large amount of hydroxyl groups. The water absorption rate for DG/MHDPE was slightly lower than that for DG/HDPE. This trend and the contact angle data were corroborative.

Water absorption data for HDPE, MHDPE, DG/HDPE, and DG/MHDPE
Conclusion
In this present study, DG/HDPE and DG/MHDPE composites were prepared and characterized, and their properties were investigated. The results showed that the addition of DG increased the hydrophilicity of the composites and improved the water absorption. From the cross-sectional morphology, it was observed that MHDPE and DG had a better interfacial compatibility; the water absorption rate for DG/MHDPE was slightly lower than that for DG/HDPE, but the mechanical properties of DG/MHDPE were improved. When the content of DG was 50%, the yield strength, crystallization temperature, and crystallinity were higher for the modified composites than for the unmodified ones and pure samples. Therefore, DG can improve the performance of materials and reduce the amount of the polymer matrix. It is a bio-filler with great potential.
References
Zarrinbakhsh N, Mohanty AK, Misra M (2015) Biocomposites from co-polypropylene and distillers’ grains. In: AIP conference proceedings. AIP Publishing, p 150005
Liu K (2011) Chemical composition of distillers grains, a review. J Agric Food Chem 59:1508–1526
Tan L, Sun Z, Zhang W, Tang Y, Morimura S, Kida K (2014) Production of bio-fuel ethanol from distilled grain waste eluted from Chinese spirit making process. Bioprocess Biosyst Eng 37:2031–2038
Tan L, Liu YH, Zhang WX (2011) Study on two-stage dilute hydrochloric acid saccharification of fermented grain waste. Food Ferment Technol 47(25-27):31
Zhang J, Zhang W-X, Wu Z-Y, Yang J, Liu Y-H, Zhong X, Deng Y (2013) A comparison of different dilute solution explosions pretreatment for conversion of distillers’ grains into ethanol. Prep Biochem Biotechnol 43:1–21
Balat M, Balat H (2009) Recent trends in global production and utilization of bio-ethanol fuel. Appl Energy 86:2273–2282
Proskurina S, Junginger M, Heinimö J, Tekinel B, Vakkilainen E (2019) Global biomass trade for energy—part 2: production and trade streams of wood pellets, liquid biofuels, charcoal, industrial roundwood and emerging energy biomass. Biofuels Bioprod Biorefin 13:371–387
Wang T-T, Wang S-P, Zhong X-Z, Sun Z-Y, Huang Y-L, Tan L, Tang Y-Q, Kida K (2017) Converting digested residue eluted from dry anaerobic digestion of distilled grain waste into value-added fertilizer by aerobic composting. J Clean Prod 166:530–536
Hao X, Benke M, Larney FJ, McAllister TA (2011) Greenhouse gas emissions when composting manure from cattle fed wheat dried c with solubles. Nutr Cycl Agroecosyst 89:105–114
Nichols JR, Schingoethe DJ, Maiga HA, Brouk MJ, Piepenbrink M (1998) Evaluation of corn distillers grains and ruminally protected lysine and methionine for lactating dairy cows. J Dairy Sci 81:482–491
Hao X, Benke MB, Li C, Larney FJ, Beauchemin KA, McAllister (2011) Nitrogen transformations and greenhouse gas emissions during composting of manure from cattle fed diets containing corn dried distillers grains with solubles and condensed tannins. Anim Feed Sci Technol 166:539–549
Ham GA, Stock RA, Klopfenstein TJ, Larson EM, Shain DH, Huffman RP (1994) Wet corn distillers byproducts compared with dried corn distillers grains with solubles as a source of protein and energy for ruminants. J Anim Sci 72:3246–3257
Leonardi C, Bertics S, Armentano LE (2005) Effect of increasing oil from distillers grains or corn oil on lactation performance. J Dairy Sci 88:2820–2827
Wu F, Munkvold GP (2008) Mycotoxins in ethanol co-products: modeling economic impacts on the livestock industry and management strategies. J Agric Food Chem 56:3900–3911
Gregorova A, Hrabalova M, Kovalcik R, Wimmer R (2011) Surface modification of spruce wood flour and effects on the dynamic fragility of PLA/wood composites. Polym Eng Sci 51:143–150
Stark NM, Matuana LM (2004) Surface chemistry changes of weathered HDPE/wood-flour composites studied by XPS and FTIR spectroscopy. Polym Degrad Stab 86:1–9
Bengtsson M, Gatenholm P, Oksman K (2005) The effect of crosslinking on the properties of polyethylene/wood flour composites. Compos Sci Technol 65:1468–1479
Tsou C-Y, Wu C-L, Tsou C-H, Chiu S-H, Suen M-C, Hung W-S (2015) Biodegradable composition of poly (lactic acid) from renewable wood flour. Polym Sci Ser B 57:473–480
Xiang Z, Watson J, Tobimatsu Y, Runge T (2014) Film-forming polymers from distillers’ grains: structural and material properties. Ind Crop Prod 59:282–289
Tsou C-H, Wu C-S, Hung W-S, De Guzman MR, Gao C, Wang R-Y, Chen J, Wan N, Peng Y-J, Suen M-C (2019) Rendering polypropylene biocomposites antibacterial through modification with oyster shell powder. Polymer (Guildf) 160:265–271
Chong MH, Chun BC, Chung Y, Cho BG (2006) Fire-retardant plastic material from oyster-shell powder and recycled polyethylene. J Appl Polym Sci 99:1583–1589
Xu Y, Wu Q, Lei Y, Yao F (2010) Creep behavior of bagasse fiber reinforced polymer composites. Bioresour Technol 101:3280–3286
Mulinari DR, Voorwald HJC, Cioffi MOH, da Silva MLC, da Cruz TG, Saron C (2009) Sugarcane bagasse cellulose/HDPE composites obtained by extrusion. Compos Sci Technol 69:214–219
Kim H-S, Yang H-S, Kim H-J, Park H-J (2004) Thermogravimetric analysis of rice husk flour filled thermoplastic polymer composites. J Therm Anal Calorim 76:395–404
Yang H-S, Kim H-J, Son J, Park H-J, Lee B-J, Hwang T-S (2004) Rice-husk flour filled polypropylene composites; mechanical and morphological study. Compos Struct 63:305–312
Tsou C-H, Hung W-S, Wu C-S, Chen J-C, Huang C-Y, Chiu S-H, Tsou C-Y, Yao W-H, Lin S-M, Chu C-K, Hu C-C, Lee K-R, Suen M-C (2014) New composition of maleic-anhydride-grafted poly (lactic acid)/rice husk with methylenediphenyl diisocyanate. Mater Sci 20:446–451
Kang DJ, Pal K, Park SJ, Bang DS, Kim J-K (2010) Effect of eggshell and silk fibroin on styrene–ethylene/butylene–styrene as bio-filler. Mater Des 31:2216–2219
Tsou C-H, Suen M-C, Yao W-H, Yeh J-T, Wu C-S, Tsou C-Y, Chiu S-H, Chen J-C, Wang RY, Lin S-M, Hung W-S, De Guzman MR, Hu C-C, Lee K-R (2014) Preparation and characterization of bioplastic-based green renewable composites from tapioca with acetyl tributyl citrate as a plasticizer. Materials (Basel) 7:5617–5632
Toro P, Quijada R, Yazdani-Pedram M, Arias JL (2007) Eggshell, a new bio-filler for polypropylene composites. Mater Lett 61:4347–4350
Tsou C-H, Yao W-H, Wu C-S, Tsou C-Y, Hung W-S, Chen J-C, Guo JP, Yuan S, Wen E, Wang R-Y, Suen M-C, Liu S-C, De Guzman MR (2019) Preparation and characterization of renewable composites from Polylactide and Rice husk for 3D printing applications. J Polym Res 26:227–237
Vao-soongnern Visit (2019) Molecular simulation of structural and dynamic properties of polymer nanoparticles composed of linear and cyclic polyethylene. Journal of Polymer Research 26(6). https://doi.org/10.1007/s10965-019-1797-2
Quiles-Díaz S, Martinez Rubi Y, Guan J, Kim KS, Couillard M, Salavagione HJ, … Simard B (2018) Enhanced Thermal Conductivity in Polymer Nanocomposites via Covalent Functionalization of Boron Nitride Nanotubes with Short Polyethylene Chains for Heat-Transfer Applications. ACS Applied Nano aterials. https://doi.org/10.1021/acsanm.8b01992
Yao Y, De Guzman MR, Duan H, Gao C, Lin X, Wen YH, Du J, Lin L, Chen J-C, Wu C-S, Suen M-C, Sun YL (2020) Infusing high-density polyethylene with graphene-zinc oxide to produce antibacterial nanocomposites with improved properties. Chinese J Polym Sci. https://doi.org/10.1007/s10118-020-2392-z
Tisserat BH, Reifschneider L, O’Kuru RH, Finkenstadt VL (2012) Mechanical and thermal properties of high density polyethylene–dried distillers grains with solubles composites. BioResources 8:59–75
Luo X, Li J, Feng J, Xie S, Lin X (2013) Evaluation of distillers grains as fillers for low density polyethylene: mechanical, rheological and thermal characterization. Compos Sci Technol 89:175–179
Wu CS, Tsou CH (2019) Fabrication, characterization, and application of biocomposites from poly(lactic acid) with renewable rice husk as reinforcement. J Polym Res 26:44–53
Jeziorska R, Szadkowska A, Zielecka M, Wenda M, Kepska B (2017) Morphology and thermal properties of HDPE nanocomposites: effect of spherical silica surface modification and compatibilizer. Polym Degrad Stab 145:70–78
Tsou C-H, Suen M-C, Tsou C-Y, Chen J-C, Yeh J-T, Lin S-M, Lai Y-C, Hwang J-Z, Huang S-H, Hung W-S (2015) Argon plasma in a new process for improving the physical and anti-bacterial properties of crosslinked cotton cellulose with dimethyloldihydroxyethyleneurea-maleic acid. Fibres Text East Eur 23(1):49–56
Essabir H, El Achaby M, Bouhfid R, Qaiss A (2015) Morphological, structural, thermal and tensile properties of high density polyethylene composites reinforced with treated argan nut shell particles. J Bionic Eng 12:129–141
Yang H-S, Kim H-J, Park H-J, Lee B-J, Hwang T-S (2006) Water absorption behavior and mechanical properties of lignocellulosic filler–polyolefin bio-composites. Compos Struct 72:429–437
Gwon JG, Lee SY, Chun SJ, Doh GH, Kim JH (2010) Effects of chemical treatments of hybrid fillers on the physical and thermal properties of wood plastic composites. Compos Part A Appl Sci Manuf 41:1491–1497
Li H-Y, Tan Y-Q, Zhang L, Zhang Y-X, Song Y-H, Ye Y, Xia M-S (2012) Bio-filler from waste shellfish shell: preparation, characterization, and its effect on the mechanical properties on polypropylene composites. J Hazard Mater 217:256–262
Luan L, Wu W, Wagner MH, Mueller M (2010) Seaweed as novel biofiller in polypropylene composites. J Appl Polym Sci 118:997–1005
Iyer KA, Torkelson JM (2014) Green composites of polypropylene and eggshell: effective biofiller size reduction and dispersion by single-step processing with solid-state shear pulverization. Compos Sci Technol 102:152–160
Essabir H, Nekhlaoui S, Malha M, Bensalah MO, Arrakhiz FZ, Qaiss A, Bouhfil R (2013) Bio-composites based on polypropylene reinforced with almond shells particles: mechanical and thermal properties. Mater Des 51:225–230
Lomakin SM, Rogovina SZ, Grachev AV, Prut EV, Alexanyan CV (2011) Thermal degradation of biodegradable blends of polyethylene with cellulose and ethylcellulose. Thermochim Acta 521:66–73
Peacock A (2000) Handbook of polyethylene: structures: properties, and applications. CRC Press
Grigoriadou I, Paraskevopoulos KM, Chrissafis K, Pvalidou E, Stamkopoulos T-G, Bikiaris D (2011) Effect of different nanoparticles on HDPE UV stability. Polym Degrad Stab 96:151–163
Singh VP, Vimal KK, Kapur GS, Sharma S, Choudhary V (2016) High-density polyethylene/halloysite nanocomposites: morphology and rheological behaviour under extensional and shear flow. J Polym Res 23:43–60
Butler MF, Donald AM, Bras W, Mant GR, Derbyshire GE, Ryan AJ (1995) A real-time simultaneous small-and wide-angle X-ray scattering study of in-situ deformation of isotropic polyethylene. Macromolecules 28:6383–6393
Kim H, Kim S, Kim HJ (2006) Enhanced interfacial adhesion of bioflour-filled poly(propylene) biocomposites by electron-beam irradiation. Macromol Mater Eng 291:762–772
Mohanty S, Verma SK, Nayak SK (2006) Dynamic mechanical and thermal properties of MAPE treated jute/HDPE composites. Compos Sci Technol 66:538–547
Tsou C-H, Lee H-T, Tsai H-A, Cheng H-J, Suen M-C (2013) Synthesis and properties of biodegradable polycaprolactone/polyurethanes by using 2, 6-pyridinedimethanol as a chain extender. Polym Degrad Stab 98:643–650
Tsou C-H, Lee H-T, De Guzman M, Tsai H-A, Wang P-N, Cheng H-J, Suen M-C (2015) Synthesis of biodegradable polycaprolactone/polyurethane by curing with H2O. Polym Bull 72:1545–1561
Tsou C-H, Lee H-T, Hung W-S, Wang C-C, Shu C-C, Suen M-C, De Guzman MR (2016) Synthesis and properties of antibacterial polyurethane with novel bis (3-pyridinemethanol) silver chain extender. Polymer (Guildf) 85:96–105
Tsou C-H, Yao W-H, Hung W-S, Suen M-C, De Guzman M, Chen J, Tsou C-Y, Wang RY, Chen J-C, Wu C-S (2018) Innovative plasma process of grafting methyl diallyl ammonium salt onto polypropylene to impart antibacterial and hydrophilic surface properties. Ind Eng Chem Res 57:2537–2545
Popescu C-M, Popescu M-C, Vasile C (2011) Structural analysis of photodegraded lime wood by means of FT-IR and 2D IR correlation spectroscopy. Int J Biol Macromol 48:667–675
Lu Y, Miller JD (2002) Carboxyl stretching vibrations of spontaneously adsorbed and LB-transferred calcium carboxylates as determined by FTIR internal reflection spectroscopy. J Colloid Interface Sci 256:41–52
Zhong ZJ (2009) Optical properties and spectroscopy of nanomaterials. World Scientific
Kakehi A, Ito S, Funahashi T, Ogasawara N (1975) Facile synthesis of 1-alkyl-2-and 4-allylidenkdihydropyri dinederivatives. Chem Lett 4:919–922
Yao W-H, Tsou C-H, Chen J-C (2016) Plasma grafting with methyl di-ally ammonium salt to impart anti-bacterial properties to polypropylene. Fibres Text East Eur 24(3):117–123
Acknowledgements
The authors would like to acknowledge the financial support from the following organizations: Wuliangye Group Co. Ltd. (CXY2019ZR001); Sichuan Province Science and Technology Support Program (2019JDRC0029); Zigong City Science and Technology (2017XC16; 2019CXRC01); Opening Project of Material Corrosion and Protection Key Laboratory of Sichuan Province (2016CL10; 2017CL03; 2019CL05; 2018CL08; 2018CL07); Opening Project of Sichuan Province, the Foundation of Introduced Talent of Sichuan University of Science and Engineering (2014RC31; 2017RCL31; 2017RCL36; 2017RCL16; 2019RC05; 2019RC07). Appreciation is also extended to the National Natural Science Foundation of China; Apex Nanotek Co. Ltd.; Ratchadapisek Sompote Fund for Postoctoral Fellowship (Chulalongkon University).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 41 kb)
Rights and permissions
About this article

Cite this article
Wen, Y., Tsou, CH., Gao, C. et al. Evaluating distillers grains as bio-fillers for high-density polyethylene. J Polym Res 27, 167 (2020). https://doi.org/10.1007/s10965-020-02148-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-020-02148-8