
Abstract
This study reports the influence of foaming temperature on morphological and thermo-mechanical characteristics of polypropylene (PP) foams prepared using two different methods of batch foaming at low saturation pressures. In the first method, involving single-step pressure-induced-foaming (PIF), the solid specimen was heated to different temperatures in a high-pressure vessel, and then saturated CO2 under pressure. Depressurization then led to foaming. In the second method, involving a two-step temperature-induced-foaming (TIF), the solid specimen was saturated with pressurized CO2 at room temperature for a specific period of time and then the CO2 laden specimen was immersed in hot glycerol bath at different temperatures for foaming. SEM micrographs of the fractured foamed specimens were employed for measurement of cell-size distribution. The average cell sizes ranged between 3 and 310 μm in specimens obtained using PIF, while in case of foams prepared by TIF, the cell sizes ranged between 30 and 70 μm. The cell sizes achieved in TIF are significantly smaller and more uniform as compared to those in PIF foams. The density (0.393–0.186 g cm−3) of PIF foams was seen to decrease with increasing foaming temperature; in contrast, in case of TIF the density remained more or less unchanged around 0.43–0.47 g cm−3 with changes in foaming temperature. The foamed specimens were characterized in uniaxial compression; the stiffness (elastic and collapse moduli) and compressive strengths of both the PIF and TIF foams were seen to decrease with increase in cell-size. The magnitude of plateau-regime stresses within the compressive stress-strain response showed strong correlation with the foam cell-wall thickness. The crystallinity of the foamed specimens was observed to decrease with increase in foaming temperatures. The thermal stability of both PIF and TIF foams in general showed improvement compared with the PP matrix.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Foams or cellular materials are porous materials -- available directly from nature (e.g. wood, cork etc.) or synthetically manufactured (e.g. styrene foams, Al foams etc.) -- that are space filled with assembly of cells with solid edges or faces [1]. Foams, offering excellent properties, material savings and cost savings, have found application in a variety of fields ranging from comfort, noise / thermal insulation, to energy-absorption applications in automotive, aerospace and personal safety [2,3,4]. Polypropylene (PP) is a versatile commodity polymer bestowed with outstanding properties and low material cost. PP is not reactive or very less reactive with environment or other compounds, which has been the most important reason for making PP based foams in industries for applications in packaging, air-filters etc. However, PP based foams are difficult to make due to the semi-crystalline nature which presents resistance to dissolution of the blowing agent into it and also due to its low melt strength which leads to rupture of foam cell walls under elongational forces during cell growth [5]. To overcome the inherent low melt-strength of PP, several PP based formulations have been developed specially for foaming [6,7,8]. Furthermore, the melt strength and extensional strain hardening of PP, and consequently its foamability, can also be increased by adding inorganic compounds such as talc and nanoparticles [5, 9,10,11,12,13,14]. The temperature of foaming is an important parameter that strongly governs cell morphology, density and other properties of the resulting foam. It is crucial to optimize the temperature and pressure settings associated with foaming, to achieve the desired foam properties.
Batch foaming is a technique that is widely adopted to carry out fundamental studies on foamability and foaming mechanisms in polymers. Batch foaming is carried out in high-pressure autoclaves, wherein a solid specimen is saturated with a physical blowing agent (such as CO2) under high pressures. For amorphous polymers, the temperature of saturation is typically kept slightly lower than the glass transition temperature. On the other hand, for semi-crystalline polymers the temperature is typically set significantly above the glass transition temperature but below the melting point. These conditions aid in increased sorption of CO2 in the polymer specimen [5]. The effect of different temperature settings on the relative density and the morphological properties of PP foams produced at constant pressure has been studied by Bao et al. [15]. By employing a saturation pressure of 25 MPa and foaming temperature range of 120–162 °C for producing foams, Bao et al. [15] reported that the density of the foams decreases with increasing foaming temperature. Similarly, Fu et al. [6] produced PP foams at high foaming pressures ranging from 8.3 to 17.3 MPa and temperature of 155 °C and reported the influence of different saturation pressures on the density of foams produced at constant saturation temperature. Prior to foaming, the solid precursors were first compressed at a temperature of 110 °C at a pressure of 414 MPa (or alternatively at 150 °C, at a pressure of 34.5 MPa). The authors observed that the increase in the saturation pressure resulted in decrease in the density of foam. They also observed a steep decrease in the density at and above a threshold pressure of 13.8 MPa, which was attributed to decrease in the crystallinity of PP at high saturation pressures. Arroyo et al. [5] studied batch foaming of PP via a modified pressure-quench method. In this method, a polymer disc placed inside a mould was employed for foaming at saturation pressures varying from 17 to 24 MPa and at a constant temperature of 155 °C. The mould used here was able to control the final density of foam. Antunes et al. [8,9,10] produced foams with PP and its nanocomposites by employing foaming pressure of 16 MPa at 135 °C. The foaming conditions and the key foam characteristics achieved in the batch foaming studies discussed in the foregoing, as well as a few other studies, are summarized in Table 1.
All these previous studies focused on solid-state batch-foaming process employing supercritical CO2 i.e., saturation was typically carried out below the melting temperature of polypropylene. In most cases, high pressures, ranging from 12.5–34.5 MPa, were employed for saturation (with the exception of Maani et al. [16] that employed lower saturation pressures of 4.5 MPa). This literature survey highlights the lack of batch-foaming studies of PP at saturation conditions that combine lower pressures (below super-critical pressure of CO2) and higher temperatures (above the melting point of PP). Furthermore, the use of temperature-induced foaming -- wherein depressurization is followed by increase in temperature to aid in cell-growth -- has not been reported in the context of PP. Temperature-induced foaming has the potential to further increase the range of temperatures employed for foaming beyond the melting point of the polymer. Therefore, the present study investigates the feasibility of producing PP foams using batch foaming at relatively lower pressures and at higher temperatures (in the vicinity of or above the melting point) by employing two foaming methodologies – a single-step pressure-induced foaming (PIF) and a two-step temperature-induced foaming (TIF). In the current study, the use of PIF at higher temperatures is aimed to more closely simulate the foaming behaviour of PP under molten conditions; this information can be very relevant to probe the mechanisms underlying industrial foaming processes especially for extruded foams.
In this study, PP foams have been produced using PIF and TIF at different processing conditions, and the resulting foams have then been evaluated for density-reduction, cell size, type of cells, and cell size-distribution. The mechanical characteristics under compression have also been studied, along with the crystallinity and thermal stability of the foams.
Experimental
Material
A commercially available PP homopolymer (Grade-M110) with MFI of 9.5 g/10 min, density of 0.912 g cm−3 and crystallinity of 40% was obtained from M/s Haldia Petrochemicals Ltd., India. CO2 gas of purity 99.9% w/w was purchased from M/s Laser gases, Delhi, India.
Sample preparation
Preparation of precursors
A micro-compounder with micro-injection jet (DSM 15, M/s Xplore) was employed to prepare solid precursor plaques from the as-received PP. For preparation of the precursors, the processing temperature was set at 190 °C with screw speed of 100 rpm, injection pressure of 0.8 MPa with a total cycle time of 9 s. The precursors of thickness 1.5 mm were prepared in two different shapes; one was a circular disc of diameter 40 mm and the other was a rectangular bar of 132 mm × 12.5 mm.
Foaming
Foams were produced by batch foaming in an autoclave (Trident Laborteck High Pressure Autoclave, diameter 75 mm, volume 1 L,) equipped with a heating device with heating capacity up to 300 °C and a cooling jacket for water circulation to control the temperature. CO2 was employed as the blowing agent for both temperature induced foaming (TIF) and pressure induced foaming (PIF).
In TIF, the solid specimen was saturated with CO2 at room temperature and a pressure of 7.0 MPa. After saturation for 24 h, the CO2 was depressurized quickly at the rate of 1.6 MPa s−1, and following a delay of 1–2 min, the gas laden specimen was dipped in hot bath of glycerol. The bath temperatures were set at different values (180 °C, 190 °C, 200 °C, 210 °C) in the various iterations. After being submerged in the glycerol bath for a about 50s, the specimen was removed and quenched in water. A similar procedure was employed by Maani et al. [16], but in their trials, the specimens were saturated at lower pressures (4.5 MPa) and the glycerine bath was maintained at a lower temperature of 155 °C.
In case of PIF, the specimens were first heated (up to temperatures ranging from 150 °C - 180 °C in various iterations) and then the molten PP specimen was pressurized with CO2 at a pressure of 7.0 MPa and saturated for 2 h. The specimens saturated at higher temperatures were cooled to 150 °C using recirculating water, before being depressurized at the rate of 1.6 MPa s−1 to induce foaming. After depressurization, the specimen was removed and kept at ambient temperature for cooling. Temperatures above 180 °C and below 150 °C were also employed for PIF trials, but at these conditions no foams were obtained. For saturation temperatures above 180 °C, material degradation was observed and only molten specimens were recovered after the foaming cycle. No foaming occurred at saturation temperatures above 180 °C. On the other hand, at temperatures below 150 °C no change was observed in the solid PP specimens within the period of saturation time under the applied pressure. Therefore, PIF results for this study have been reported only for temperatures ranging from 150 to 180 °C. It needs to be noted here that the temperatures employed for PIF in the current study are in the vicinity of the melting point of the PP, and are significantly higher than those employed in similar studies reported by others [6, 15,16,17,18,19]. The experimental setups of the PIF and TIF processes are schematically depicted Fig. 1.

Experimental setup a Pressure induced foaming, b Temperature induced foaming
Characterization
Density
The mass density of foamed samples was measured by a balance (M/s Mettler Toledo) equipped with a density measurement kit as per ASTM D792 [6]. The density was calculated by weighing the sample in air and water, and by using formula [6]:
Where ρf is density of foam sample, a is mass of sample in air, b is mass of sample completely immersed in water. The relative density Rρ is defined as the ratio of density of foam (ρf) to the density of unfoamed solid (ρs):
The volume expansion ratio is, Rv, is defined as the reciprocal of relative density Rρ of the foam:
Porosity
The void fraction or porosity (ϕ) was calculated using the values of bulk density of the foam, ρf, the density of the physical blowing agent, ρb and the density of the solid unfoamed polymer, ρs using the relationship shown below.
Since ρb < < ρs, and ρb < < ρf, Eq. (4) can be re-written as [20]:
Morphological characterization
The cell morphologies of the foams were characterized using scanning electron microscopy (SEM) (EVO MA15 M/s Carl Zeiss). Prior to microscopic analysis, the foamed specimens were cryogenically fractured in liquid nitrogen and the fractured specimens were sputter coated with gold (Emitech SC7620, Quorum Technologies Ltd., UK). The SEM micrographs of the fractured surfaces were then analysed using ImageJ software to survey the morphology. For each specimen surface, 4 SEM images were analysed (resulting in total surveyed areas ranging from 0.46 × 10−3 – 90.95 × 10−3 cm2 for different micrographs). For each micrograph analysed, firstly, the perimeter of each cell in the SEM image was identified automatically using ImageJ edge-detection algorithms and then from the perimeter, the diameter of each cell was calculated assuming an equivalent circular cell. The cell-sizes were then sorted from the smallest to largest. The difference between the smallest and largest sizes was divided into multiple equi-size-interval bins, and the number of cells lying within each bin was quantified to arrive at a size distribution. From this size distribution, the volume-averaged diameter of all the cells measured from the micrographs, Dv, was calculated as:
Where n is the number of cells, di is perimeter equivalent diameter of each counted cell.
Mechanical properties
The mechanical behaviour of foams produced by TIF and PIF was measured in the uniaxial compression mode using a universal testing machine (Instron – 3382, UK) at room temperature under quasi-static conditions, employing a velocity of 1 mm min−1. The uniaxial compressive loading was stopped at strains at which either the specimens showed edge failure, or when the stress-strain curve showed a sharp upturn indicative of densification – whichever occurred earlier.
Differential scanning calorimetry
The thermophysical properties and crystallinity of foams were measured with differential scanning calorimetry (DSC Q20, TA Instruments, USA). The measurements were carried out in nitrogen environment using a temperature ramp from 30 °C to 200 °C with heating rate of 10 °C min−1. Crystallinity in the specimen was quantified by comparing the enthalpy of melting from the DSC curve with the enthalpy of melting for 100% crystalline PP which is taken as 207 J g−1 [21, 22].
Thermogravimetric analysis
The thermal degradation and stability characteristics of the foams were measured using thermogravimetric analyser (TGA Q50, TA Instruments, USA). The measurement was conducted in nitrogen environment using a temperature ramp from 30 °C to 500 °C with a heating rate of 10 °C min−1 to examine the relative mass loss of the samples.
Results and discussion
The foaming conditions and process parameters for the different iterations of PIF and TIF are listed in Tables 2 and 3 respectively, along with the measured densities of the resulting foams. As discussed earlier, relative density is defined as the ratio of density of foam to that of the unfoamed material and so the relative density of PP-00 specimen (the unfoamed PP matrix) is 1. It can be seen from Table 2 that the density of the foam shows a decreasing trend with increasing temperature in PIF. On the other hand, at different conditions of TIF, there was no significant variation in the density of the resulting foams.
From this study, the lower end of densities achieved is in the range of 0.2 g cm−3 which is higher than those achieved by [6, 8, 15] – this could be due to a lower level of initial sorption of gas due to the lower pressures employed. The higher end of densities however, is consistently below 0.45, which is significantly lesser than that achieved by [6, 15] and equivalent to that achieved in [8,9,10]. It should be noted that all these prior studies referred here employ significantly higher pressures compared to this study.
The porosity of foams was calculated from Eq. (5), and as such it is correlated with the relative density of the foams. It needs to be noted here that no independent measurement of porosity was carried out in this study, and therefore the porosities reported for the foams follow the exact inverse trend as the relative densities of the foams. From Table 2 for PIF, it can be seen that as the relative density of the foam decreases with increasing saturation temperature, the calculated porosity of the foams also showed a corresponding increase. On the other hand, from Table 3 for TIF, it can be observed that the relative density shows an increase upon increase of temperature from 180 to 190 °C, and then stays more or less unaltered at higher temperatures (within experimental error). Correspondingly, the calculated porosity shows a decline going from 180 to 190 °C and does not change thereafter at higher foaming temperatures.
The trends in density and porosity of the foams are next correlated with the observations of morphological characteristics of the foams in the subsequent.
Morphology and cell size distribution
The SEM micrographs of the PIF foams produced under various conditions listed in Table 2 are shown in Figs. 2 and 3. The cell size distribution quantified from the analysis carried out on the images in Fig. 2 have been depicted in Fig. 4.

SEM micrographs of PP foams prepared using PIF as per conditions listed in Table 2 a PPF-01, b PPF-02, c PPF-03, d PPF-04

SEM micrograph of PP foams prepared using pressure induced foaming at 180 °C showing details of the size gradient of the cells at the edge of the specimen

Cell size distribution measured from SEM micrographs of PP foams prepared using PIF at a 150 and 160 °C, and b at 170 and 180 °C. The distributions are presented in two different panels due to the large scale-difference in cell sizes obtained at the various temperatures
As may be seen from the panels in Fig. 2, PIF at all temperatures resulted in relatively broad distribution of cell sizes. A qualitative inspection of the figures also reveals a pronounced variation of cell size with the increase in PIF temperature. Further, it may be qualitatively concluded from the micrographs that the foams achieved at all the temperatures are predominantly closed-cell; this may be inferred from the presence of cell-walls around each individual cells, except a few instances which show cell continuity and coalescence. It needs to be noted here that no independent measurement of the closed-cell content of the foams was carried out.
A preliminary comparison of the micrographs from the different specimens also establishes consistency with the quantified densities and porosities of the specimens. Going from PPF-01 to PPF-03, an increase in cell size can be clearly observed, and is consistent with the progressively reducing density (corresponding increase in porosity) reported in Table 2. Going from PPF-03 to PPF-04, a decrease in cell sizes can be qualitatively inferred, however the cell density is seen to increase. A combination of these factors seems to have resulted in the achievement of densities in PPF-04 that is not significantly different from that of PPF-03.
These qualitative trends are further clear from inspection of the cell size distributions obtained from the image analyses of the micrographs, plotted in Fig. 4. From Fig. 4, while noting the broad distribution in cell sizes, a clear trend emerges of increasing cell size with increase in the temperature of PIF up to 170 °C, as the size distributions show pronounced and progressive shift towards the right (larger cell sizes) with increase in temperature. The average bubble size for PIF at the three temperatures calculated from these distributions are 3.0 μm at 150 °C (PPF-01), 4.0 μm at 160 °C (PPF-02), and 312.0 μm at 170 °C (PPF-03). Such a significant increase in the bubble size with increase in temperature from 160 °C to 170 °C may be attributed to the decrease in viscosity of the polymer at increased temperature resulting in reduced resistance to cell growth [23]. In addition, a lower viscosity as a result of an increase in temperature may also lead to cell collapse.
However, upon further increasing the temperature to 180 °C in PIF, the cell sizes are seen to decrease; this is obvious from a left shift of the size distribution in Fig. 4b. The average cell diameter was calculated to be 115 μm at 180 °C (PPF-04). The foam produced by PIF at 180 °C shown in Fig. 3 reveals a pronounced gradient in cell sizes, going from the edge to the middle; the cells on the edge have smaller size as compared to the middle. Based on the marked deviation in foam morphology at 180 °C, which is not in line with the trend observed from 150 to 170 °C, it may be argued that at 180 °C, there is a limiting temperature condition for PIF for the pressures employed. Also, at 180 °C and beyond, the solubility of CO2 in PP matrix has started to significantly decrease while the diffusivity out of PP has increased, resulting in poor sorption and retention of CO2 at the pressure employed in this study. These effects may be exacerbated at higher temperatures further explaining the lack of foam formation in specimens subjected to PIF higher than 180 °C [24].
The SEM micrographs of the TIF foams produced under various conditions listed in Table 3 are shown in Figs. 5 and 6. The cell size distributions quantified from the image analyses carried out on the micrographs in Fig. 5 have been depicted in Fig. 7.

SEM micrographs of PP foams prepared using temperature induced foaming as per conditions listed in Table 3 a PPF-05, b PPF-06, c PPF-07, d PPF-08

SEM micrographs of PP foams prepared using temperature induced foaming with details depicting collapse of bubbles at high temperature

Cell size distribution measured from SEM micrographs of PP foams prepared using temperature induced foaming, under various conditions listed in Table 3
From a preliminary inspection of the micrographs in Fig. 5, it may be qualitatively concluded that the TIF foams achieved at all the temperatures are predominantly closed-cell; this may be inferred from the presence of clear boundaries of unfoamed PP matrix around each individual cells, except a few instances which show cell continuity and coalescence. Once again, it needs to be noted here that no independent measurement of the closed-cell content of the foams was carried out.
A preliminary comparison of the micrographs from the different TIF specimens also establishes consistency with the quantified densities and porosities of the specimens. In PPF-05, a significantly higher number density of cells is clearly inferred compared to the specimens produced at higher temperature. The specimens produced at the three higher temperatures do not show any marked differences in morphology. These qualitative observations are consistent with the trends in density of the specimens, wherein PPF-05 had the lowest density, while the PPF-06, PPF-07 and PPF-08 specimens produced at higher foaming temperatures had similar densities which were all higher than that of the PPF-05 specimen.
A quick comparison of Figs. 2 and 5 reveals stark contrasts between the morphology of foams achieved from PIF and TIF.
Firstly, the regions comprised of the PP matrix, separating the foam cells, are much thinner in PIF compared to those in TIF. In case of PIF, these PP regions are uniformly thin and may be qualified as cell walls. On the other hand, in case of TIF the solid PP regions between the cells are too thick to be qualified as cell walls and rather these regions appear to be “islands” of unfoamed matrix within the foamed structure.
Secondly, the cell densities - number of cells per unit area, assessed qualitatively through visual inspection of the micrographs - in PIF, especially at the lower temperatures, are significantly higher compared to those achieved in TIF. Even in case of the samples with lower cell densities (e.g., PIF at 170 °C), the PP cell-walls bounding the foam cells are very thin, implying a uniform volumetric expansion of the foam with growth in the cell size. On the other hand, a decreased cell density while increasing the temperature from 180 °C to 190 °C in TIF, there is a corresponding increase in the solid PP regions in the micrographs, indicating non-uniform growth of the cells.
These qualitative contrasts point to marked differences in the cell nucleation and growth mechanisms in the two routes implemented during the foaming process. In case of PIF, a high nucleation density, combined with a more uniformly distributed cell growth seems to have resulted in large volumetric expansions accompanying foaming. On the other hand, in case of TIF, a high nucleation density (obvious in the micrograph at 180 °C) is followed by selective growth of only a small portion of the cells resulting in regions that may be completely devoid of cells. The selective growth of a few bubbles is clearly visible in TIF at higher temperatures. Further, at these temperatures, it is possible that a significant amount of dissolved gas desorbs, while the larger bubbles grow at the expense of smaller bubbles.
Another clear contrast between PIF and TIF is in terms of the trend in sizes of the largest cells achieved with the change in foaming temperature. In case of PIF, upon varying the temperature from 150 to 170 °C, the sizes of the largest cells grow almost twenty-fold (with a decrease in expansion ratio observed at 180 °C as discussed in the foregoing). On the other hand, the sizes of the largest cells achieved in TIF was relatively constant at around 100–200 μm at all the temperatures.
Further, in case of TIF foams produced at 200 °C and 210 °C (PPF-07 and PPF-08) the bubbles appear collapsed and merged as shown in Fig. 6. This collapsing and merging of bubbles make the cells uneven considerably increase the size of bubbles.
The above details are also obvious in the quantified cell size distributions for TIF foams plotted in Fig. 7. As mentioned earlier, the increase in temperature does not significantly alter the maximum cell-sizes, and the cell-size distributions for the various temperatures overlap. The larger cell sizes achieved through bubble collapse and coalescence at higher temperatures is also obvious from the tails of the size distributions obtained for PPF-07 and PPF-08.
From these distributions, the average cell sizes were calculated as 32 μm, 69 μm, 71 μm and 73 μm for PPF-05, PPF-06, PPF-07 and PPF-08 respectively.
To summarize the foregoing discussions, the trends in the average cell size calculated from the measured distributions with respect to the various processing temperatures have been plotted in Fig. 8. As mentioned earlier, PIF foams show a more pronounced dependence of cell size on temperature compared to TIF foams. Also, as discussed earlier, the significantly larger cell-sizes, achieved in PIF combined with thinner PP cell-walls are indicative of more uniform and higher overall volumetric expansion during foaming. This is reflected in the trends of measured foam densities listed in Tables 2 and 3 and discussed at the beginning of this section.

Variation of average cell size with foaming temperature for pressure induced foams and temperature induced foams
The decrease in density of PIF foams with increasing temperature is consistent with the corresponding observed increase of cell sizes as plotted in Fig. 8. It needs to be reiterated that the density of PPF-04 remains similar to PPF-03 despite the significant reduction in cell sizes (as seen in Fig. 8), due to the significantly higher cell number density (cf. Fig. 2c and d) especially at the specimen edges (cf. Fig. 3). As far as temperature induced foaming is concerned, the density does not vary much with temperature and it ranges between 0.431–0.472 g cm-3, consistent with the overlapping cell-size distributions. The slight increase in density in going from 180 °C to 190 °C is consistent with the significantly higher cell number density observed at 180 °C (see Fig. 5a) compared to that at 190 °C. As discussed earlier, for TIF foams produced at higher temperatures, even though the average cell size is almost twice that achieved at 180 °C (see Fig. 8), the foam density increases rather than decreases due to significant reduction in number of cells.
Mechanical characteristics in uniaxial compression
The stress-strain responses of the unfoamed PP matrix as well as the various PIF and TIF foams in uniaxial compression are plotted in Fig. 9.

Compressive stress-strain plot of PP matrix and foams
From the stress-strain response of the foams shown in Fig. 9, three distinct regimes may be inferred; (i) a linear regime at low strains, (ii) a plateau regime at intermediate strains, where the compressive stress remains more or less constant with increase in strains and (iii) another linear regime with increased slope at higher strains, indicative of densification.
The maximum compressive strain achievable in the tests and the stress corresponding to this maximum compressive strain, are listed in Table 4, along with two measures of foam stiffness. The homogenized elastic modulus of the foam is estimated from the initial linear–elastic region of the compressive stress-strain curves. The collapse modulus or collapse strength is defined as the intersection point of linearized curve of elastic regime and the linearized curve of the plateau regime [25].
Generally, the mechanical behaviour of polymeric foams not only depends on the mechanical properties of matrix but also on the physical and structural properties of foams such as density, cell size and cell size distribution [26, 27]. It is clear from an inspection of Fig. 9 as well as Table 4 that the elastic modulus, collapse modulus and the ultimate compressive strength of the foams may be roughly ordered in the same way as the foam densities.
However, the differences in magnitudes of these parameters between the PIF and TIF foams can be more strongly correlated to the differences in the cell-sizes achieved in these foams. Larger cell sizes with typically thinner cell walls in PIF foams correspond to significantly lower stiffness measures as compared to TIF foams which have more pronounced regions of unfoamed matrix separating the foam cells. These observations are consistent with similar trends observed in prior studies (see e.g., Refs [28, 29]).
The clear ordering of the stress-strain curves, especially the stresses in the plateau-regime, with the foam density is another clear qualitative indicator of the closed-cell nature of the foams. For closed cell foams, the stresses in plateau-regime (which involves bending as well as contraction of the foam cell walls) are mainly governed by the pressure build up within the enclosed gas within the foamed cells [30, 31]. The TIF foams achieve significantly higher plateau stresses compared to the PIF foams. The higher plateau stresses in TIF foams can be correlated with the thicker regions of unfoamed matrix which separate the foam cells (cf. Fig. 5) which can sustain much higher pressure build-up within the cells. On the other hand, the significantly lower plateau stresses for PIF foams, especially for PPF-03 and PPF-04 specimens is consistent with the observation of very thin cell walls (cf. Fig. 2c and d) which can fail at low stress levels.
Crystallinity
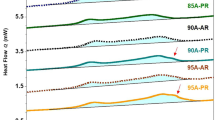
The DSC heat flow plots for the virgin PP and the various foamed specimens are shown in Fig. 10. The melting point (Tm) of the specimens was calculated from the heating curves (Fig. 10a) and the crystallization temperature (Tc) was calculated from the cooling curves (Fig. 10b). The Tm of the unfoamed PP matrix was found to be 168 °C whereas in case of the foams, the melting points were observed to lie between 167 and 170 °C. The Tc of unfoamed PP was observed at 123.6 °C and for foams, Tc ranged between 121 and 125 °C.

Heat Flow rates measured during a DSC temperature ramp experiment carried out on neat PP and the foamed specimens a melting curves and b crystallization curves
The crystalline fraction of the PP foams, determined using Eq. 7, has been plotted as a function of the foaming temperature in Fig. 11. It can be observed that the crystallinity of PP foams decreases with increase in saturation temperature in PIF process. At lower temperatures of PIF below melting point, the foam growth would involve high degree of solid-state orientation in the PP, which is consistent with the high observed crystallinity for these specimens. At higher temperatures of PIF, even though there is pronounced cell growth and the PP cell walls are thinned (Fig. 2c and d), there does not seem to be significant chain orientation in the PP regions which might be attributed to the reduced crystallinity in the specimens produced at higher temperatures.

Crystallinity of the foamed specimens as a function of Temperature of foaming
In case of TIF, while all the four foaming temperatures were above the melting point of PP, it may be argued that the specimens may not have uniformly attained the bath temperature, and therefore for the specimens achieved at the lower temperatures (180 °C, 190 °C) the foam growth may have been accompanied by PP orientation. This may be particularly true for the specimen at 180 °C, which shows a much more pronounced cell density, which is possibly due to high crystallinities in these samples. Further, the reduction in crystallinity at 210 °C can be due to the fact that at higher temperature, PP may be in molten state during foam growth which restricts the orientation of matrix chains. Of course, the same phenomenology should have resulted in a lower crystalline fraction for the TIF foam specimen prepared at 200 °C also, but that is not the case. The factors underlying these subtle differences are not fully understood and warrant further investigations.
Thermal stability
Thermal behaviour of the virgin matrix and its foams are illustrated in the TGA/dTG thermographs given in Fig. 12a and b respectively. The temperature corresponding to a weight loss of 5% is defined as the initial degradation temperature (IDT) and is used as thermal stability indicator for the developed foams. The data, including the temperature corresponding to the 5%, 50% of weight loss and residual weight, are listed in Table 5. It is clear that all the foams have single step degradation evolving between 280 °C to 470 °C. From Fig. 12a, it is clear that compared to the unfoamed PP matrix, all the foams (with the exception of PPF-03 and PPF-04) display a higher initial degradation temperature and also overall better thermal stability. In case of PIF foams, the degradation temperatures show a clear inverse correlation with the cell size; i.e., foams with larger cell size have a lower IDT. Whether or not the entrapped CO2 has a role to play in affecting the thermal stability of the foam needs to be further investigated. No clear correlation may be observed between thermal stability and cell-size in case of TIF foams.

Thermal properties of PP foams a Thermogravimetric weight loss, b first derivative (dTG) curves
Conclusion
PP foams were successfully produced from a commercial PP resin using batch-foaming with CO2 at different temperatures by two methods; pressure-induced foaming (PIF) and temperature-induced foaming (TIF). Both foaming routes employed a saturation pressure of 7.0 MPa, which was significantly lower than the pressures employed in prior batch foaming studies. Further, the temperatures employed for PIF in the current study are in the vicinity of the melting point of the PP, and are significantly higher than those employed in analogous prior studies [6, 15,16,17,18,19]. In the current study, the use of PIF at higher temperatures is aimed to more closely simulate the foaming behaviour of PP under molten conditions so as to better probe the mechanisms underlying industrial foaming processes, especially extruded foams. In comparing the trends obtained in the current study with those the prior studies employing batch foaming, the following points may be highlighted:
-
From this study, the lower end of densities achieved is in the range of 0.2 which is higher than those achieved by [6, 8, 15] – this could be due to a lower level of initial sorption of gas due to the lower pressures employed.
-
The higher end of densities however, is consistently below 0.45, which is significantly less than that achieved by [6, 15] and equivalent to that achieved in [8,9,10]. It should be noted that all these references employ significantly higher pressure compared to this study.
-
Higher temperatures employed in this study push the foam cell size to larger values which are consistent with the observations of Antunes et al. [10].
-
This study thus provides an alternative route of achieving low density foams using lower pressure batch foaming combined with high temperatures.
The detailed assessment of correlations between the density, porosity, morphology and thermomechanical properties of the foams, may be summarized as below:
The density (0.393–0.186 g cm-3) of foams decrease with increasing the temperature in PIF. By contrast, in TIF the density is maintained around 0.43–0.47 g cm-3 with increasing the temperature. The different trends in density, combined with the stark qualitative differences in foam morphology, point to marked differences in the cell nucleation and growth mechanisms in the two routes for the foam. In case of PIF, a high nucleation density, combined with a more uniformly distributed cell growth seems to have resulted in large volumetric expansions accompanying foaming. On the other hand, in case of TIF, a high nucleation density is followed by selective growth of only a small portion of the cells resulting in regions that may be completely devoid of cells.
It was also observed that the cells in TIF processed at higher temperatures collapse and merged with the adjacent cells. However, in PIF process the cells do not collapse with increasing temperature, rather they stretch and become larger due to low melt strength property of polypropylene. Both these observations are consistent with reduced crystallinity of the foamed specimens obtained at the highest temperatures for PIF and TIF. By contrast, at the lower end of temperatures investigated both for TIF and PIF, foam growth appears to be accompanied by PP matrix stretch and orientation, resulting in higher observed crystallinities in the foamed specimens.
Qualitative assessment of morphology as well as the strong correlation between the foam density and the stiffness, plateau stresses and compressive strength of the foams clearly imply a predominantly closed-cell nature of the foams. The stiffness and strength are inversely correlated with the size of the foam cells, while the plateau stresses are directly correlated with the thickness of the cell-walls between the cells.
The thermal stability of the foamed specimens is observed to be in general better compared to unfoamed PP. Specimens with very large cell sizes however seem to have lower degradation temperatures compared to unfoamed PP. The correlations between thermal stability, cell size and the physical blowing agent chemistry need to be investigated further.
References
Gibson LJ, Ashby MF (1997) Cellular solids: Structure and properties, vol 2E. Cambridge University Press, Cambridge. https://doi.org/10.1017/CBO9781139878326
Eaves D (2004) Handbook of polymer foams, 1st ed. Rapra Technology Ltd, UK
Mills NJ (2007) Polymer foams handbook: engineering and biomechanics applications and design guide. Butterworth-Heinemann
Degischer HP, Kriszt B (2002) Handbook of cellular metals - production, processing, applications. Handbook of Cellular Metals Wiley-VCH. https://doi.org/10.1002/3527600558
Saiz-Arroyo C, De Saja JA, Velasco JI, Rodríguez-Pérez MÁ (2012) Moulded polypropylene foams produced using chemical or physical blowing agents: structure-properties relationship. J Mater Sci 47:5680–5692
Fu D, Chen F, Kuang T, Li D, Peng X, Chiu DY, Lin CS, Lee LJ (2016) Supercritical CO2 foaming of pressure-induced-flow processed linear polypropylene. Mater Des. Elsevier Ltd 93: 509–513
Antunes M, Gedler G, Abbasi H, Velasco JI (2016) Graphene nanoplatelets as a multifunctional filler for polymer foams. Materials Today: Proceedings 3:S233–S239
Antunes M, Realinho V, Ardanuy M, Maspoch ML, Velasco JI (2011) Mechanical properties and morphology of multifunctional polypropylene foams. Cell Polym 30:187–200
Lobos J, Velankar S (2014) How much do nanoparticle fillers improve the modulus and strength of polymer foams? J Cell Plast 0:1–32
Antunes M, Gedler G, Velasco JI (2013) Multifunctional nanocomposite foams based on polypropylene with carbon nanofillers. J Cell Plast 49:259–279
Zhai W, Park CB, Kontopoulou M (2011) Nanosilica addition dramatically improves the cell morphology and expansion ratio of polypropylene heterophasic copolymer foams blown in continuous extrusion. Ind Eng Chem Res 50:7282–7289
Sorrentino L, Aurilia M, Cafiero L, Iannace S (2011) Nanocomposite foams from high-performance thermoplastics. J Appl Polym Sci 122:3701–3711
Luo Y, Xin C, Zheng D, Li Z, Zhu W, Wu S, Zheng Q, He Y (2015) Effect of processing history on the rheological properties, crystallization and foamability of branched polypropylene. J Polym Res 22:1–13
Laguna-Gutierrez E, Lopez-Gil A, Saiz-Arroyo C, Van Hooghten R, Moldenaers P, Rodriguez-Perez MA (2016) Extensional rheology, cellular structure, mechanical behavior relationships in HMS PP/montmorillonite foams with similar densities. J Polym Res 23
Bao JB, Nyantakyi Junior A, Weng GS, Wang J, Fang YW, Hu GH (2016) Tensile and impact properties of microcellular isotactic polypropylene (PP) foams obtained by supercritical carbon dioxide. J Supercrit Fluids. Elsevier BV 111: 63–73
Maani A, Naguib HE, Heuzey M-C, Carreau PJ (2013) Foaming behavior of microcellular thermoplastic olefin blends. J Cell Plast 49:223–244
Xu ZM, Jiang XL, Liu T, Hu GH, Zhao L, Zhu ZN, Yuan WK (2007) Foaming of polypropylene with supercritical carbon dioxide. J Supercrit Fluids 41:299–310
Wong A, Guo Y, Parka CB (2013) Fundamental mechanisms of cell nucleation in polypropylene foaming with supercritical carbon dioxide - effects of extensional stresses and crystals. J Supercrit Fluids. Elsevier B.V. 79: 142–151
Velasco JI, Antunes M, Realinho V (2010) Foaming behaviour, structure, and properties of polypropylene nanocomposites foams. J Nanomater 2010:1–11
Istrate OM, Chen B (2011) Relative modulus–relative density relationships in low density polymer–clay nanocomposite foams. Soft Matter 7:1840–1848
Faye A, Leduc M, Brisson J (2014) Crystallization control of etherethersulfone copolymers by regular insertion of an allyl functionality. Polym Chem 5:2548
Wal A, Van Der Mulder JJ, Gaymans RJ (1998) Fracture of polypropylene : the effect of crystallinity. Polymer 39:5477–5481
Bao JB, Liu T, Zhao L, Hu GH (2011) A two-step depressurization batch process for the formation of bi-modal cell structure polystyrene foams using ScCO2. J Supercrit Fluids. Elsevier B.V. 55: 1104–1114
Li D, Liu T, Zhao L, Yuan W (2009) Solubility and diffusivity of carbon dioxide in solid-state isotactic polypropylene by the pressure - decay method. Ind Eng Chem Res 48:7117–7124
Wan F, Tran M-P, Leblanc C, Béchet E, Plougonven E, Léonard A, Detrembleur C, Noels L, Thomassin J-M, Nguyen VD (2015) Experimental and computational micro-mechanical investigations of compressive properties of polypropylene/multi-walled carbon nanotubes nanocomposite foams. Mech Mater. Elsevier Ltd 91: 95–118
Chen L, Schadler LS, Ozisik R (2011) An experimental and theoretical investigation of the compressive properties of multi-walled carbon nanotube / poly ( methyl methacrylate ) nanocomposite foams. Polymer. Elsevier Ltd 52: 2899–2909
Saha MC, Mahfuz H, Chakravarty UK, Uddin M, Kabir ME, Jeelani S (2005) Effect of density, microstructure, and strain rate on compression behavior of polymeric foams. Mater Sci Eng A 406:328–336
Kuncir EJ, Wirta RW, Golbranson FL (1990) Load-bearing characteristics of polyethylene foam: an examination of structural and compression properties. J Rehabil Res Dev 27:229–238
Moscoso-Sanchez FJ, Mendizabal E, Jasso-Gastinel CF, Ortega-Gudino P, Robledo-Ortiz JR, Gonzalez-Nunez R, Rodrigue D (2015) Morphological and mechanical characterization of foamed polyethylene via biaxial rotational molding. J Cell Plast 51:489–503
Vries DVWM de (2009) Characterization of Polymeric Foams. Master’s Thesis (MT 09.22) Eindhoven University of Technology
Iba H, Nishikawa Y, Urayama K (2016) Nonlinear stress-strain behavior of elastomer foams investigated by various types of deformation. Polymer. Elsevier Ltd 83: 190–198
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kumar, A., Patham, B., Mohanty, S. et al. Effect of temperature on thermal, mechanical and morphological properties of polypropylene foams prepared by single step and two step batch foaming process. J Polym Res 26, 80 (2019). https://doi.org/10.1007/s10965-019-1699-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-019-1699-3