
Abstract
In this study poly(2-ethyl-2-oxazoline) (PEOXA) chains that contains 11 mol% of benzophenone molecules was synthesized and coated on either 3-ethoxybenzophenonesilane-modified inorganic or bare organic substrates. Upon irradiation under UV light, the photo-active benzophenone molecules enabled the formation of polymer network as well as attachment of the polymer network onto the substrates. Important variables for the generation of hydrogel film with high gel content and stability, such as the heat treatment for solvent removal, the UV wavelength (that determines the irradiation energy), and the input of energy dose were varied and their influence to the gel content and stability of the hydrogel film was studied. The thickness, lifetime of benzophenone, and chemical composition of the film were determined using ellipsometry, UV/Vis spectroscopy, and XPS methods, respectively. On a film that has been exposed to physiological buffer for 14 days, XPS results indicated that chemical degradation of the copolymer did not take place. Ellipsometry results, however, indicated that some portion of the film detached and the remaining thickness was dependent on the input of energy dose during the hydrogel preparation. It was shown that when suitable conditions are applied during preparation, a stable surface-attached PEOXA-based hydrogel, i.e. approximately 78% gel content and 75–90% stability after 30 days of incubation in physiological buffer, could be generated on the surface. Dry and swollen thicknesses of the stable surface-attached film measured from AFM experiments revealed a swelling factor of 1.7. Furthermore, the AFM morphology image showed a homogenous polymer film with an average roughness of 30 nm. Protein adsorption test revealed that the resulting surface-attached PEOXA-based hydrogel film on PMMA substrate hinders BSA adsorption to the same extent as the reference system generated from benzophenone-bearing poly(dimethylacrylamide) (PDMAA).
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introductions
Surface fouling on biomedical devices is ubiquitous, cost-demanding, and can be life-threatening [1]. Coating of biomedical devices with non-fouling film has so far been one of the most attractive strategies in the prevention of surface fouling [1]. Some pre-requisites for polymers that can be used for the fabrication of non-fouling surface coatings are hydrophilicity (the polymers should be hydrophilic), charge (the polymers should carry net neutral charge), and Hydrogen-bond groups (the polymers should contain H-bond donor but should not contain H-acceptor groups) [2,3,4,5]. Among a broad spectrum of polymers that fulfill the mentioned prerequisites are poly(ethylene glycol) [6,7,8], phosphorylcholine [9, 10], polysaccharide [11,12,13], poly(vinylpyrrolidone) [14], poly(vinyl alcohol) [15], polyacrylamides [5, 16] and polyacrylates [17], and the poly(2-oxazoline)s [18, 19]. For the latest, there has been a significant emergence of this class of polymer since the first reports on poly(2-oxazoline)s (POXs) around 5 (five) decades ago due to their excellent biocompatibility and stealth behavior, in which they do not readily trigger foreign body response [20]. The reason for this may come from the similarity between POXs and peptides in terms of chemical structure, in which they consist of Carbon-Carbon-Nitrogen at their main chains. Poly(2-methyl-2-oxazoline) (PMOXA) for example, it offers isomerism to poly(homo-alanin) peptide chain. The peptide-like structure offers not only biocompatibility, but also resistance to auto-oxidation and thus high stability [21, 22]. Surface grafting of POXs have been performed on different inorganic and organic substrates using various methods to modulate their interfacial, physico-chemical properties and provide functions to the modified surface. In particular, the performance of POX coatings as biointerfaces to hinder nonspecific biological adhesion has been extensively explored [23]. Grafted poly(2-methyl-2-oxazoline) (PMOXA) significantly reduced non-specific adsorption of serum proteins on Nb2O5 surfaces [24] and increased stability of DNA particles against serum proteins and enzymatic DNase-I digestion [21]. Good transfection efficiency combined with low cytotoxicity of the PMOXA-grafted DNA particles was demonstrated in COS-7 cells [21]. In comparison to PEG brush film, the golden standard in the area, PMOXA brush film was significantly more stable upon long term exposure to biological environment [22, 25]. Morgese and Benetti [23] reviewed a plethora of grafting strategies of PAOXAs onto surfaces, and concluded that poly(2-alkyl-2-oxazoline)s (PAOXAs) have been one of the most promising materials among the possible solutions for biocompatible, non-fouling surface coatings [23]. Among the grafting strategies are the reaction between catechol-PAOXAs and metal oxides [26], xanthane-PAOXAs and citrate-Au [27], alkyne-PAOXAs and thiol-Au [28], thiol-PAOXAs and carboxyl-polyorganosiloxane [29], thiol-PAOXAs and Au [30], alkyne-PAOXAs and azide-polystyrene [31], as well as amino-PAOXAs and cyanuric chloride-lipid [32]. The strategy has also encompassed PAOXA-containing block or graft copolymers presenting segments or co-monomer functions showing a strong affinity for the surface, as well as precoating of the surface with polymers or grafting promotors that display a high density of functional groups that can react with PAOXAs [33,34,35]. In the development of biocompatible hydrogels, Sramkova et al. [36] reported the preparation of poly(2-oxazoline) network using the specific thiol-ene “click” reaction between dithiol groups carried by the crosslinkers and alkene groups carried by the poly(2-oxazoline) chains. While the above-mentioned strategies require specific reactions between the functional groups presented on the surface (and/or at the crosslinker in the case of polymer network) and their counterparts attached on the PAOXA chains, a simpler and convenient surface photo-grafting as well as photo-crosslinking of PAOXAs has been reported. The grafting steps include surface functionalization with a photo-reactive compound such as benzophenone [37] and prefluorophenyl azide (PFPA) [38], spin-coating of PAOXA, and UV illumination that triggers the formation of carbon radical and nitrene, respectively, capable of binding the spin-coated PAOXAs without the need of any specific coupling functions [23], in which benzophenone generally results in higher crosslinking yields compared to PFPA [39]. Interestingly, poly(2-ethyl-2-oxazoline) (PEOXA) thin (monolayer) film prepared using benzophenone-mediated grafting strategy did not show any substantial reduction of protein adsorption on glass surfaces [37]. Furthermore, Dhende et al. [40] developed a PEOXA-based polymer network for antimicrobial surface coating applications. The resulting network, however, carries high positive charge, not satisfying the prerequisites for non-fouling surface coatings. In the present work, a non-fouling film of poly(2-oxazoline)-based polymer network (hydrogel) on surfaces is developed by partially functionalizing poly(2-ethyl-2-oxazoline) (PEOXA) chains with benzophenone units that react with any C-H bonds in the vicinity upon UV illumination and allow for a direct and simple immediate attachment to the substrate, regardless whether it is a (hydrophobically-modified) metal or metal oxide, or a polymer. The polymer network thus covalently bounds to the surfaces of the substrates and have a much higher thickness and robustness compared to monolayer.
Material and methods
Materials
All chemicals were purchased from Sigma-Aldrich, unless otherwise stated. The biotinylated-laminin and Cy5-streptavidin were purchased from Cytoskeleton, Inc. and VWR, respectively.
Synthesis of poly(2-ethyl-2-oxazoline)-m%ethyleneimine (PEOXA-m%EI)
The synthesis was performed following a previously published protocol [40]. Poly(2-ethyl-2-oxazoline) (PEOXA) with Mw ~ 50,000 g/mol, DP ~ 500, and PDI ~ 3–4 was purchased from Sigma-Aldrich (CAS 25805–17-8, catalog no. 372846 ALDRICH). PEOXA (5 g) and HCl (16.8%, 105 ml) were added to a 250-ml round-bottom flask. The mixture was stirred and refluxed at 100 °C. After 40 min., the mixture was cooled down to room temperature. Then, neutralization using KOH solution was performed until neutral pH (~7) is reached. The neutral solution was transferred to a dialysis membrane with MWCO ~ 14,000 g/mol (Carl Roth, catalog no. 1784.1) and dialyzed against aquadest bath for at least 2 × 24 h, with changing of the aquadest bath at least every 24 h. The solution was freeze-dried until (white) PEOXA-m%EI polymer powder is obtained. Characterization was performed using 1H-NMR spectroscopy.
Synthesis of poly(2-ethyl-2-oxazoline)-m%ethyleneiminebenzophenone (PEOXA-m%EIBP)
The synthesis was performed following a previously published protocol [40]. PEOXA-m%EI (4 g, 4.7 mmol EI monomer) and tert-amyl alcohol (20 ml) were added to a 50-ml round-bottom flask and stirred until the polymer powder is dissolved. Then, K2CO3 (0.85 g) and 4-(bromomethyl) benzophenone 96% (1.36 g) were added to the mixture. The mixture was stirred and heated up to 95 °C. After an overnight reaction, the mixture was cooled down to room temperature, and the solvent, tert-amyl alcohol, was exaporated using a rotary vaporizer. The (yellowish, oily) solid residue was re-dissolved in dichloromethane. The PEOXA-m%EIBP polymer was then precipitated in excess amount of n-hexane. After vacuum-filtration, the polymer was collected and vacuum-dried for several days to remove traces of dichloromethane and n-hexane. Characterization was performed using 1H-NMR spectroscopy.
Film thickness, chemical composition, and lifetime of benzophenone molecules
The copolymer was spin-coated (600 rpm, 100 s) from a 50 mg/ml solution in ethanol onto a 1.5 × 1.5 cm Silicon (Si) wafer, previously modified using 3-ethoxybenzophenonesilane, for both film thickness and XPS measurements. The film thickness was then measured using ellipsometry and atomic force microscopy (AFM) methods, while the chemical composition on the Si wafer surface was measured using XPS. The same copolymer solution was spin-coated on a quartz substrate for benzophenone lifetime measurement. From time to time between 0 to 132 min, the sample on quartz substrate sample was taken out from the Stratalinker® and the change in absorbance intensity was monitored using UV/Visible spectroscopy.
Gel content and stability test
Following the spin-coating step as described above, illumination under UV light was performed using Stratalinker® UV crosslinker. The thickness of the polymer film on top of the Si wafer was then measured using either ellipsometry or AFM (thickness t0). After rinsing with ethanol and blow-drying under Nitrogen, the wafer was incubated in PBS buffer for 24 h. The wafer was then taken out from the PBS solution, thoroughly washed with aquadest and ethanol, and blow-dried under Nitrogen. The film thickness was measured again using either ellipsometry or AFM (thickness t1). The gel content was calculated according to Eq. 1.
The stability test was performed using the same protocol as the gel content measurement, however, with prolonged incubation time of the wafer in PBS buffer, up to 30 days. The film thickness was measured from time to time, resulting thickness data tn, where n is 1, 2, 3, … 30 days. The remaining thickness was calculated according to Eq. 2.
Swelling factor
The copolymer was spin-coated (600 rpm, 100 s) from a 50 mg/ml solution in ethanol onto a 1.5 × 1.5 cm Si wafer, and illuminated using UV light. The polymers that are not crosslinked were extracted with ethanol. The remaining wafer surface-attached film was then incubated in PBS buffer for 14 days, rinsed carefully with extensive amount of water and, subsequently, ethanol, and dried under nitrogen flow. Immediately after drying, the dry thickness of the remaining film was measured using AFM. Afterwards, the wafer was placed into a small polystyrene plate followed by addition of PBS to soak the film, and the swollen thickness of the film on top of the wafer was measured under the PBS environment. The swelling factor was estimated by comparing the swollen (tswollen) and the dry (tdry) thickness, according to Eq. 3.
Protein adsorption test
The copolymer was dip-coated (speed 100 mm/s) from a 50 mg/ml solution in ethanol onto a poly(methylmethacrylate) (PMMA) slide substrate (75x25x1.0 mm, PMMA Mitsubishi Shinkolite A farblos “version 1800”). After crosslinking with UV light at 365 nm, the slide was incubated in PBS buffer for at least 24 h, followed by ultrasonication for 5 min, rinsing with aquadest and ethanol, and blow-drying under Nitrogen. A plastic frame that could accommodate 65 μl liquid was glued on top of the PMMA slide, with an arranged position so that the border between copolymer-coated and bare surface was in the middle of the frame. Biotinylated bovine serum albumin (biotin-BSA, 0.1 mg/ml, 65 μl) was placed inside the frame, and the protein adsorption was allowed to take place for 2 h. The slides were then thoroughly rinsed, incubated in PBS buffer, and placed in a shaker for at least 15 min. The rinsing-and-washing step was done for 2 (two) times. After blow-drying under Nitrogen, cyanine 5-bearing streptavidin (cy5-stretavidin, 0.001 mg/ml, 65 μl) was placed inside the frame, and the reaction between biotin and streptavidin was allowed to proceed for 30 min. The same rinsing-and-washing protocol as described above was then repeated. The red-fluorescence signal from the cy5 dye was then measured by means of fluorescence reader. ImageJ software was used for the quantitative analysis of the signal intensity (presented in Gray mean value).
Results and discussions
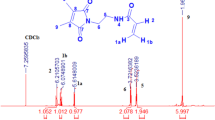
The strategy described here employs poly(2-ethyl-2-oxazoline) (PEOXA) with an approximately 50,000 g/mol molecular mass. Controlled partial hydrolysis of the polymer results in PEOXA with controlled ethylenimine (EI) content along its chain (PEOXA-m%EI). This functionality serves as the coupling sites for the incorporation of benzophenone (BP) units through a reaction between the secondary amine groups and bromide groups attached on the 4-(bromomethyl) BP units. This reaction results in PEOXA-m%EIBP copolymer. As a control, a reaction between PEOXA and 4-(bromomethyl) BP is also performed. Figure 1a shows the NMR spectra of 4-(bromomethyl) BP and PEOXA that are not covalently linked to each other due to the absence of secondary amine functionality. Figure 1b, on the other hand, shows a successful coupling between the 4-(bromomethyl) BP and PEOXA-m%EI that results in a final product PEOXA-m%EIBP. In this study PEOXA-11%EIBP is employed. This copolymer consists of 2-ethyl-2-oxazoline (EOXA, 89 mol%) as the main component that serves as a hydrophilic matrix to prevent non-specific interactions with biological environment. The second component is ethyleniminebenzophenone units (EIBP, 11 mol%) that provide the photo-reactive functionality, i.e. upon UV light illumination, the benzophenone units can covalently bind to any C-H bonds in the vicinity through C,H-insertion crosslinking (CHiC) reactions. The presence of residual secondary amine group, protonated-, and quaternized-amine groups, if any, cannot be detected from the NMR spectra.

NMR spectra of (a) unreacted 4-(bromomethyl) benzophenone and PEOXA, b PEOXA-11%EIBP, where x ∼ 445 and y ∼ 55
Following the synthesis and NMR characterization, the (co)polymer is immobilized on the substrate surfaces and PEOXA-based hydrogel is generated. One of important parameters in controlling the process and rating the quality of hydrogels (polymer networks) is the gel content. To this, an approximately 1000 nm-thick of copolymer is spin-coated onto Si wafers that are pre-functionalized with 3-ethoxybenzophenonesilane. Several process parameters during the preparation of the network are then varied and the gel content is determined. In purification step, the copolymer is dissolved in dichloromethane and precipitated in n-hexane. The NMR characterization (Fig. 1) shows that residual dichloromethane (CH2Cl2) and n-hexane (C6H14) solvents cannot be totally removed from the copolymer powder. In addition, coating the copolymer onto the substrates employs ethanol as the PEOXA-11%EIBP’s solvent. Such organic solvent molecules can also react through C,H-insertion reaction, but this will not connect two copolymer chains and thus not result in increase of the crosslink density, an important factor that influences the gel content. Evaporating the residual solvents that are “trapped” in the coated film before triggering the crosslinking reaction will eliminate this problem. Figure 2a shows that heating treatment between 5 and 10 min at 80 °C does increase the gel content of the PEOXA-11%EIBP film. Longer heating treatment does not significantly change the gel content anymore. Based on this finding, a 10-min heating treatment is always applied to the coated film on the surface during any preparation of PEOXA-11%EIBP network in this entire study. As controls, PEOXA and PEOXA-11%EI film are included in the investigation, in which both show a low gel content value in comparison to PEOXA-11%EIBP film. This result demonstrates the role of benzophenone molecules along the polymer chains in forming a thick copolymer network of PEOXA-11%EIBP on the surface.

The influence of (a) preheating treatment at 80 °C (UV light at 365 nm) and b the UV light wavelength to the gel content of PEOXA-based hydrogel films
Using the photon energy equation, E = h.c/λ, where E is the photon energy, h is the Planck’s constant, c is the velocity of light, and λ is the wavelength of light, one can estimate that the energy irradiated by UV light is higher at lower wavelength. Higher irradiation energy activates BP units faster than the lower one [41]. However, it is also known that certain UV light energy can degrade organic materials including polymers [42]. Figure 2b shows that using an identical total energy dose of either 2 or 4 J/cm2, illumination under UV light at 254 nm results in a film with significantly lower gel content compared to that under UV light at 365 nm. Despite the advantage that the activation of BP units is faster at 254 nm, the lower gel content indicates that the former is more degrading than the latter [41]. The less degrading UV light at 365 nm, is then used in the further study. Moreover, together with the results from UV-Vis experiments, the gel content of the film as a function of energy dose upon UV light illumination at 365 nm is presented in Fig. 3b (further discussion below).

a Absorbance change in UV/Vis spectra of benzophenone in PEOXA-11%EIBP film as a function of UV light exposure time at 365 nm and b the gel content and normalized absorbance of benzophenone as a function of UV light energy dose at 365 nm. The thickness of the film during UV irradiation was approximately 1000 nm
Upon irradiation with UV light, the reaction progress of benzophenone, in which new C-C bonds are formed, can be monitored indirectly by following the decrease in the π-π* transition of benzophenone at around 260 nm [40]. To this, an approximately 1000 nm-thick PEOXA-11%EIBP film is spin-coated on a quartz surface, heat treated at 80 °C for 10 min, and illuminated under UV light at 365 nm. From time to time, the sample is taken from the crosslinker apparatus and the absorbance is measured using UV/Vis spectroscopy. Figure 3a shows the time dependent changes in the absorption spectra of the benzophenone-bearing PEOXA polymeric film upon UV light irradiation. It is seen that the absorbance maximum decreases with increasing irradiation time. All benzophenone units on the surface react after 132 min of UV light irradiation (in our apparatus, this equals to approximately 18 J/cm2 energy dose at 365 nm). The absorbance maximum at each time is then identified. After subtraction with a baseline estimated from the data followed by normalization with the highest maximum, the normalized absorbance of benzophenone, together with the gel content, is plotted as a function of energy dose in Fig. 3b. It is seen that the gel content increases from 0 to 78% with increasing energy dose up to 4 J/cm2 and with decreasing normalized absorbance of benzophenone (which means increasing reacted BP in the film) down to 0.55, then plateaues. Higher energy dose and more reacted BP in the film does not increase the gel content anymore from 78% value. This means that from a 1000-nm thick spin-coated polymer film, approximately 780 nm is firmly attached on the substrate surface while the rest of the film is leached away during extraction. This relatively low gel content value compared to that of other hydrogel system using poly(dimethylacrylamide) (up to 99%) [43] may be originated from the fact that the PEOXA-based films contain some impurities that are not covalently crosslinked (only physically trapped) within the network. These impurities are then washed away from the substrate surface during extraction after the CHiC process. The gel content value of PEOXA-11%EIBP in the present work is, however, in an excellent agreement with the gel content value of a similar system that has been reported by Dhende and co- workers [40], in which the authors reported an approximately 80% gel content from an initially 93-nm thick polymer film before CHiC reaction.
XPS experiments are then performed to obtain chemical information of the modified surfaces. The XPS samples are prepared following the same procedure as described in the gel content measurements, using an energy dose of 4 J/cm2 at 365 nm. Figure 4a, b, c, and d show the survey spectra of bare Si wafer, spin-coated PEOXA-11%EIBP film before any further treatments, spin-coated PEOXA-11%EIBP film after CHiC process and extraction, and spin-coated PEOXA-11%EIBP film after CHiC process, extraction, and incubation in PBS, respectively. Compared to the spectra of bare Si wafer in Fig. 4a, the domination of C 1 s and N 1 s signals, as well as disappearance of Si 2 s and Si 2p peaks on the spectra of PEOXA-11%EIBP-coated wafer in Fig. 4c and d demonstrates the formation of a stable surface-attached copolymer film that provides a sufficient “shielding” for the Si surface. In agreement with the above-described explanation on gel content and impurities, Fig. 4b shows the presence of (minor) bromide-containing species, originated from the synthesis step, on the PEOXA-11%EIBP-coated surface before any extraction and incubation of the substrate. The bromide signals, however, disappear after extraction and incubation steps (Fig. 4c and d). It is worthwhile to note that the incubation in PBS buffer for 14 days is performed to test the film stability, since exposure to physiological environment during application may lead to copolymer degradation and/or detachment of the copolymer film from the surface. Elemental analysis on the surface enables the determination if the copolymer undergoes any degradation reactions that change its chemical composition. Based on the chemical structure of PEOXA-11%EIBP shown in Fig. 1, the theoretical C, N, and O atomic concentrations exclusively originate from the copolymer are 76, 12, and 12%, respectively, while the corresponding values based on the XPS spectra in Fig. 4d are 75, 13, and 12%. The similar (almost identical) theoretical and experimental atomic concentration values indicates that copolymer degradation is unlikely in this context. In literature, it has been reported that surface-attached poly(2-methy-2-oxazoline) in brush configuration is chemically stable upon exposure to HEPES buffer [22]. The XPS data in the present work are thus in agreement with the literature and validate the chemical stability of poly(2-oxazoline) upon exposure to physiological buffer.

The survey XPS spectra of (a) bare Si wafer, b PEOXA-11%EIBP on Si wafer after spin-coating, c PEOXA-11%EIBP on Si wafer after UV light irradiation and extraction using PEOXA-11%EIBP-dissolving solvent, and d PEOXA-11%EIBP on Si wafer after UV light illumination, extraction, and 14-days incubation in PBS buffer. The C, N, and O atomic concentrations obtained from spectra (d) calculated from the integrated area of C1s, N1 s, and O1s peaks are 75, 13, and 12%, respectively
As another measure of stability, ellipsometry experiments are performed and the remaining thicknesses after a controlled period of film incubation in PBS are obtained. The film is prepared using 3 (three) different energy doses for the CHiC reaction. Figure 5 shows that the energy dose used during the copolymer network preparation indeed plays role in the film stability, in which the stability increases with increasing energy dose. Interestingly, at the same periods of incubation, extreme lower remaining thicknesses are observed when the CHiC is performed at 2 J/cm2 energy dose compared to both at 4 and 6 J/cm2. The percolation theory studied by Koerner et al. [43] may explain the phenomenon observed at 2 J/cm2. The authors stated that during illumination under UV light, the percolating clusters grow from the top through the polymer film until it reaches and reacts with a cluster with connection to the substrate. An infinite network in the direction orthogonal to the surface has to be formed, in order to avoid detachment/removal of all (loosely connected) material on top of the substrate-bound clusters during extraction and/or incubation in any dissolving media. Presumably, 2 J/cm2 energy dose is not sufficient to form such a network, resulting in an unstable connection between the material on the top and the substrate. The loosely connected material is then easily detached upon long exposure to the PBS buffer. Increasing the energy dose to 4 or 6 J/cm2 apparently increases the chance for the infinite network formation inside the film and eliminates this problem. Only slight difference of stability is observed between the film prepared using 4 J/cm2 compared to film prepared using 6 J/cm2 energy dose. From Fig. 5 it can be summed up that when ≥4 J/cm2 UV light energy dose is used for the CHiC reactions, 75–90% of the PEOXA-11%EIBP film, relative to the film after gel content measurement, remains on the substrate surface after incubation in physiological buffer PBS for up to 30 days. This means that from a 1000-nm thick spin-coated polymer film, approximately 780 nm remains on the substrate surface after extraction with polymer dissolving solvents, as described previously in the gel content discussion part. From the remaining 780-nm film, 10–25% detaches during incubation in physiological buffer for up to 30 days, leaving an approximately 585–702-nm film on the surface. It is worthwhile to note that our experiments with initial polymer film thickness other than 1000 nm shows that the relative gel content and remaining thickness values are independent on the initial polymer film thickness after spin-coating.

The remaining thickness of PEOXA-11%EIBP film relative to the film thickness after gel content measurement (approximately 780 nm) as a function of incubation period in PBS buffer and of UV light energy dose (at 365 nm) used for the CHiC reaction in the preparation of polymer network. The film shows increasing stability with increasing energy dose
As previously mentioned, an important prerequisite for a non-fouling film in the context of biomedical devices is the hydrophilicity of the film. In this study, the dry and swollen thickness of PEOXA-11%EIBP film on Si wafer is measured using AFM after 14 days of incubation in PBS buffer. A swelling factor is then determined to demonstrate the film hydrophilicity. The topographic images, measured height profiles, and surface morphology from AFM measurements are presented in Fig. 6a, b, c, and d. It is seen that the swollen and dry thickness values are approximately 660 and 390 nm, respectively. According to eq. 3, these values result in a swelling factor of 1.7, which is in an excellent agreement with the reported swelling factor of surface-attached poly(dimethylacrylamide) with similar benzophenone molar concentration along the polymer chain (10%) [44]. The influence of crosslinker content to the swelling behavior of surface-attached hydrogel has been discussed in the literature [44, 45]. In the present study the hydrophobic nature of benzophenone molecules should render the PEOXA chains less hydrophilic, and thus less swelling capability. In addition, a high crosslinking density leads to a more compact polymer network and restricted movement of polymer chains, preventing the penetration of water molecules into the network [45]. In Fig. 6d, the surface morphology image shows the presence of a satisfactorily homogenous PEOXA-11%EIBP film on the Si wafer, with an average roughness 30 nm.

The AFM data of PEOXA-11%EIBP film after extraction of non-crosslinked materials from the surface followed by incubation of the polymer network in PBS buffer for 14 days. a, b The topographic images in swollen and dry state, respectively. c The measured height profiles obtained by tracing the arbitrary lines in the corresponding images shown in (a and b). The average height (thickness) values for swollen and dry film are 660 and 390 nm, respectively, resulting in a swelling factor of 1.7. d The surface morphology image of the polymer network in dry state, i.e. corresponds to the polymer film shown in image (b)
Following the physical and chemical characterizations, the non-fouling properties of the PEOXA-based hydrogel film is investigated. To this, the copolymer is dip-coated onto a poly(methylmethacrylate) (PMMA) slide from a 50 mg/ml solution in ethanol, illuminated under UV light at 365 nm with 4 J/cm2 energy dose, extracted with ethanol, and incubated in PBS buffer for 2 days (this incubation period is chosen because it is seen in Fig. 5 that the remaining thickness during stability test plateaus after 2 days of incubation). After additional careful washing and drying, the surface is exposed to biotin-bearing BSA proteins. The biotin functionality enables the reaction with Cy5-bearing streptavidin. The fluorescence signal from the Cy5 on the surface is then measured and presented in Fig. 7. As controls, PDMAA-based hydrogel film, reported as a highly non-fouling surface coating [46], and bare PMMA slide are included in the protein adsorption test. The PDMAA-based hydrogel film is prepared from PDMAA-5%MABP copolymers using identical procedure as used for the preparation of PEOXA-11%EIBP film. It is seen in Fig. 7 that modification of bare PMMA surfaces with either PDMAA- or PEOXA-based hydrogel renders the surface highly non-fouling.

The qualitative (inzet) and quantitative fluorescence signal intensity on different surfaces after exposure to biotin-BSA and cy5-streptavidin. Comparable to PDMAA-based hydrogel, the PEOXA-based hydrogel film significantly reduces BSA protein adsorption on PMAA surface, demonstrating its non-fouling properties
Summary and conclusion
The study aimed at developing surface-attached hydrogel film based on poly(oxazoline) that shows non-fouling properties. PEOXA chains with desired content of ethleneimine benzophenone (EIBP) groups were sythesized and used for substrate modification. The benzophenone units acted as photocrosslinkers to form PEOXA-based polymer networks. When substrates such as 3-ethoxybenzophenonesilane-modified silicon (Si) wafer and bare PMMA slide were coated with the polymer and illuminated with UV light, the photoactive groups were activated and they initiated the reaction with carbon-hydrogen bonds nearby through C,H-insertion. Through this procedure surface-attached PEOXA-based hydrogel film was generated. The surfaces of the substrates could thus be modified and transformed into non-fouling surfaces with this method. The results showed that it was important to remove all organic solvent molecules from the film as organic molecules could also participate in the C,H-insertion reactions. A short heat treatment of the copolymer film before starting the CHiC reaction thus increased the gel content of the film. It was also found that both irradiation energy and energy dose of the UV light were important for the gel content and film stability. Too high irradiation energy (such as that irradiated by UV light at 254 nm) resulted in polymer degradation and gel content decrease. UV light at higher wavelength such as at 365 nm was thus recommended for the PEOXA-based film. However, a sufficient energy dose input was required for generating a stable film. When the energy dose was too low, insufficient crosslinking was formed and the stability of the films diminished upon longer exposure to physiological buffer PBS that also acted as dissolving media for the copolymer. Preceding the investigation of non-fouling properties, the water holding capacity of the hydrogel was identified based on its swelling factor. It was found that the hydrogel swells by a factor of 1.7, which is in a good agreement with similar hydrogel system based on PDMAA. The morphology image showed a homogenous dry polymer network film on the substrate surface. At the end of the study, the non-fouling properties of the stably surface-attached PEOXA-based hydrogel on PMMA substrates was investigated by means of immunostaining method. It was found that surfaces modified with PEOXA-based hydrogel repeled protein adsorption to the same extent as the reference system, i.e. surfaces modified with PDMAA-based hydrogel. The approach to develop a non-fouling PEOXA-based hydrogel film described here was very easy to perform without the need of any sophisticated equipments, rendering the technique to be an attractive technology. This study provides important parameters values in the development of the PEOXA-based hydrogel system.
References
Li J, Taylor M, Zhang Z (2017) In: Zhang Z, Wagner V (eds) Anti-fouling medical coatings. Springer, Cham
Chapman RG, Ostuni E, Liang MN, Meluleni G, Kim E, Yan L, Pier G, Warren HS, Whitesides GM (2001) Polymeric thin films that resist the adsorption of proteins and the adhesion of Bacteria. Langmuir 17(4):1225–1233
Hoffmann J, Groll J, Heuts J, Rong H, Klee D, Ziemer G, Moeller M, Wendel HP (2006) Blood cell and plasma protein repellent properties of Star-PEG-modified surfaces. J Biomater Sci Polym Ed 17(9):985–996
Otsuka H, Nagasaki Y, Kataoka K (2004) Characterization of aldehyde-PEG tethered surfaces: influence of PEG chain length on the specific biorecognition. Langmuir 20(26):11285–11287
Mahdavi H, Norouzian S (2018) Preparation and characterization of modified ultrafiltration nylon 6 membrane modified by poly (acrylamide-co-maleic anhydride). J Polym Res 25(10):222
Krsko P, Libera M (2005) Biointeractive hydrogels. Materials Today (Oxford United Kingdom) 8(12):36–44
Koh W-G, Revzin A, Simonian A, Reeves T, Pishko M (2003) BioMEMs materials and fabrication technology: control of mammalian cell and bacteria adhesion on substrates micropatterned with poly(ethylene glycol) hydrogels. Biomed Microdevices 5(1):11–19
Park JH, Bae YH (2003) Hydrogels based on poly(ethylene oxide) and poly(tetramethylene oxide) or poly(dimethyl siloxane). II. Physical properties and bacterial adhesion. J Appl Polym Sci 89(6):1505–1514
Lewis AL (2000) Phosphorylcholine-based polymers and their use in the prevention of biofouling. Colloids Surf B Biointerfaces 18(3,4):261–275
West SL et al (2003) The biocompatibility of crosslinkable copolymer coatings containing sulfobetaines and phosphobetaines. Biomaterials 25(7–8):1195–1204
Li P, Wang J, Lu WC, Sun H, Huang N (2005) Surface characterization and antibacterial evaluation of poly(ethylene terephthalate) modified by chitosan-immobilization. Key Eng Mater 288-289(Advanced Biomaterials VI):331–334
An YH, Farino M, Kang QK, Demcheva MV, Vournakis J (2005) Glucosamine coating for inhibiting bacterial adhesion to titanium surfaces. Key Eng Mater 288-289(Advanced Biomaterials VI):343–346
Morra M, Cassineli C (1999) Non-fouling properties of polysaccharide-coated surfaces. J Biomater Sci Polym Ed 10(10):1107–1124
Tunney MM, Gorman SP (2002) Evaluation of a poly(vinyl pyrrolidone)-coated biomaterial for urological use. Biomaterials 23(23):4601–4608
Koziarz J, Yamazaki H (1999) Stabilization of polyvinyl alcohol coating of polyester cloth for reduction of bacterial adhesion. Biotechnol Tech 13(4):221–225
Chen H, Chen Q, Hu R, Wang H, Newby BMZ, Chang Y, Zheng J (2015) Mechanically strong hybrid double network hydrogels with antifouling properties. J Mater Chem B 3(27):5426–5435
Chen H, Zhao C, Zhang M, Chen Q, Ma J, Zheng J (2016) Molecular understanding and structural-based Design of Polyacrylamides and Polyacrylates as antifouling materials. Langmuir 32(14):3315–3330
Hoogenboom R (2007) Poly(2-oxazoline)s: alive and kicking. Macromol Chem Phys 208(1):18–25
Hoogenboom R (2009) Poly(2-oxazoline)s: a polymer class with numerous potential applications. Angew Chem Int Ed 48(43):7978–7994
Hoogenboom R (2017) 50years of poly(2-oxazoline)s. Eur Polym J 88:448–450
von Erlach T, Zwicker S, Pidhatika B, Konradi R, Textor M, Hall H, Lühmann T (2011) Formation and characterization of DNA-polymer-condensates based on poly(2-methyl-2-oxazoline) grafted poly(l-lysine) for non-viral delivery of therapeutic DNA. Biomaterials 32(22):5291–5303
Pidhatika B, Rodenstein M, Chen Y, Rakhmatullina E, Mühlebach A, Acikgöz C, Textor M, Konradi R (2012) Comparative stability studies of poly(2-methyl-2-oxazoline) and poly(ethylene glycol) brush coatings. Biointerphases 7(1):1–15
Morgese G, Benetti EM (2017) Polyoxazoline biointerfaces by surface grafting. Eur Polym J 88:470–485
Pidhatika B, Möller J, Benetti EM, Konradi R, Rakhmatullina E, Mühlebach A, Zimmermann R, Werner C, Vogel V, Textor M (2010) The role of the interplay between polymer architecture and bacterial surface properties on the microbial adhesion to polyoxazoline-based ultrathin films. Biomaterials 31(36):9462–9472
Chen Y, Pidhatika B, von Erlach T, Konradi R, Textor M, Hall H, Lühmann T (2014) Comparative assessment of the stability of nonfouling poly(2-methyl-2-oxazoline) and poly(ethylene glycol) surface films: An in vitro cell culture study. Biointerphases 9(3):031003
Morgese G, Causin V, Maggini M, Corrà S, Gross S, Benetti EM (2015) Ultrastable suspensions of Polyoxazoline-functionalized ZnO single nanocrystals. Chem Mater 27(8):2957–2964
de la Rosa Victor R et al (2015) Colorimetric logic gates based on poly(2-alkyl-2-oxazoline)-coated gold nanoparticles. Adv Funct Mater 25(17):2511–2519
Mansfield EDH, de la Rosa VR, Kowalczyk RM, Grillo I, Hoogenboom R, Sillence K, Hole P, Williams AC, Khutoryanskiy VV (2016) Side chain variations radically alter the diffusion of poly(2-alkyl-2-oxazoline) functionalised nanoparticles through a mucosal barrier. Biomater Sci 4(9):1318–1327
Koshkina O, Lang T, Thiermann R, Docter D, Stauber RH, Secker C, Schlaad H, Weidner S, Mohr B, Maskos M, Bertin A (2015) Temperature-triggered protein adsorption on polymer-coated nanoparticles in serum. Langmuir 31(32):8873–8881
Zheng X, Zhang C, Bai L, Liu S, Tan L, Wang Y (2015) Antifouling property of monothiol-terminated bottle-brush poly(methylacrylic acid)-graft-poly(2-methyl-2-oxazoline) copolymer on gold surfaces. J Mater Chem B 3(9):1921–1930
Lind JU, Acikgöz C, Daugaard AE, Andresen TL, Hvilsted S, Textor M, Larsen NB (2012) Micropatterning of functional conductive polymers with multiple surface chemistries in register. Langmuir 28(15):6502–6511
Tauhardt L, Frant M, Pretzel D, Hartlieb M, Bücher C, Hildebrand G, Schröter B, Weber C, Kempe K, Gottschaldt M, Liefeith K, Schubert US (2014) Amine end-functionalized poly(2-ethyl-2-oxazoline) as promising coating material for antifouling applications. J Mater Chem B 2(30):4883–4893
Quintana R, Gosa M, Jańczewski D, Kutnyanszky E, Vancso GJ (2013) Enhanced stability of low fouling Zwitterionic polymer brushes in seawater with Diblock architecture. Langmuir 29(34):10859–10867
Divandari M, Dehghani ES, Spencer ND, Ramakrishna SN, Benetti EM (2016) Understanding the effect of hydrophobic protecting blocks on the stability and biopassivity of polymer brushes in aqueous environments: a Tiramis√π for cell-culture applications. Polymer 98:470–480
Pidhatika B, Möller J, Vogel V, Konradi R (2008) Nonfouling surface coatings based on poly(2-methyl-2-oxazoline). CHIMIA Int J Chem 62:264–269
Šrámková P, Zahoranová A, Kroneková Z, Šišková A, Kronek J (2017) Poly(2-oxazoline) hydrogels by photoinduced thiol-ene “click” reaction using different dithiol crosslinkers. J Polym Res 24(5):82
Chang B-J, Prucker O, Groh E, Wallrath A, Dahm M, Rühe J (2002) Surface-attached polymer monolayers for the control of endothelial cell adhesion. Colloids Surf A Physicochem Eng Asp 198-200:519–526
Wang H, Li L, Tong Q, Yan M (2011) Evaluation of Photochemically immobilized poly(2-ethyl-2-oxazoline) thin films as protein-resistant surfaces. ACS Appl Mater Interfaces 3(9):3463–3471
Dorman G, Prestwich GD (1994) Benzophenone photophores in biochemistry. Biochemistry 33(19):5661–5673
Dhende VP, Samanta S, Jones DM, Hardin IR, Locklin J (2011) One-step photochemical synthesis of permanent, nonleaching, ultrathin antimicrobial coatings for textiles and plastics. ACS Appl Mater Interfaces 3(8):2830–2837
Riga KE et al (2017) On the limits of benzophenone as cross-linker for surface-attached polymer hydrogels. Polymers 9(12)
Cui H, Hanus R, Kessler MR (2013) Degradation of ROMP-based bio-renewable polymers by UV radiation. Polym Degrad Stab 98(11):2357–2365
Körner M, Prucker O, Rühe J (2016) Kinetics of the generation of surface-attached polymer networks through C, H-insertion reactions. Macromolecules 49(7):2438–2447
Li K et al (2015) On the lubrication mechanism of surfaces covered with surface-attached hydrogels. Macromol Chem Phys 217(4):526–536
Nakhjiri MT, Bagheri Marandi G, Kurdtabar M (2018) Effect of bis[2-(methacryloyloxy)ethyl] phosphate as a crosslinker on poly(AAm-co-AMPS)/Na-MMT hydrogel nanocomposite as potential adsorbent for dyes: kinetic, isotherm and thermodynamic study. J Polym Res 25(11):244
Pandiyarajan CK, Prucker O, Zieger B, Rühe J (2013) Influence of the molecular structure of surface-attached poly(N-alkyl acrylamide) coatings on the interaction of surfaces with proteins, cells and blood platelets. Macromol Biosci 13(7):873–884
Acknowledgements
We gratefully acknowledge the Alexander von Humboldt foundation for financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Pidhatika, B., Zhao, N. & Rühe, J. Development of surface-attached thin film of non-fouling hydrogel from poly(2-oxazoline). J Polym Res 26, 21 (2019). https://doi.org/10.1007/s10965-018-1677-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-018-1677-1