
Abstract
Novel flexible dielectric composites composed of polyvinyl alcohol (PVA), polyethylene glycol (PEG), and graphene oxide (GO) with high dielectric constant and low dielectric loss have been developed using facile and eco-friendly colloidal processing technique. The structure and morphology of the PVA/PEG/GO composites were evaluated using Fourier transform infrared spectroscopy (FTIR), Raman spectroscopy, UV-vis spectroscopy (UV-vis), X-ray diffraction (XRD), scanning electron microscopy (SEM), and atomic force microscopy (AFM). The dielectric behavior of PVA/PEG/GO composites was investigated in the wide range of frequencies from 50 Hz to 20 MHz and temperature in the range 40 to 150 °C using impedance spectroscopy. The dielectric constant for PVA and PVA/PEG (50/50) blend film was found to be 10.71 (50 Hz, 150 °C) and 31.22 (50 Hz, 150 °C), respectively. The dielectric constant for PVA/PEG/GO composite with 3 wt% GO was found to be 644.39 (50 Hz, 150 °C) which is 60 times greater than the dielectric constant of PVA and 20 times greater than the dielectric constant of PVA/PEG (50/50) blend film. The PVA/PEG/GO composites not only show high dielectric constant but also show low dielectric loss which is highly attractive for practical applications. These findings underline the possibilities of using PVA/PEG/GO composites as a flexible dielectric material for high-performance energy storage applications such as embedded capacitors.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
With the continuous advancement of modern electronics, the demand and importance of eco-friendly multifunctional and flexible dielectric materials with excellent dielectric properties have become more stringent. Among these, lightweight materials with high dielectric constant and ultra low dielectric loss have aroused tremendous research enthusiasm because of their manifold applications such as high-k gate dielectrics, high charge storage devices, actuators, artificial muscles, and electronic packaging [1–4]. The main challenge in designing such dielectric devices especially those for energy storage are the simultaneous achievement of a high dielectric constant as well as low dielectric loss. Polymer-based dielectrics are the suitable candidate for energy storage applications because of their excellent properties such as high flexibility, easy processability, low dielectric loss, high electric breakdown strength, and low cost [5–7]. However, despite innumerable merits, some polymers have very low dielectric constant (k < 5) which is not sufficient for energy storage application. Thus, increasing the dielectric constant of polymers is the key issue for practical applications while retaining their other excellent properties. Several strategies have been developed in order to improve the dielectric constant of neat polymers [8–10]. Two approaches are proven to be efficient in making polymer-based dielectrics more desirable for energy storage application. While the first approach is the introduction of high-k ceramic fillers into the polymer matrix [11–15], fabricating percolative polymer composites using conducting fillers such as carbon black, carbon fibers, carbon nanotubes, exfoliated graphite nanoplatelets, fullerenes, nanodiamonds, and metal nanoparticles comes as the second. However, the increment in the dielectric properties usually results in the decrement of mechanical properties of the polymer composites [16] for the ceramic fillers and in the form of greater dielectric loss values for the conductive fillers. In the latter case, since high dielectric constants are often obtained especially at filler concentrations close to percolation threshold, interlayers or shells have been introduced within the conducting fillers to prevent their direct contact and to avoid leakage of current and thus dielectric loss [17, 18].
Among the carbon fillers, graphene has shown tremendous potential for synthesis of polymer nanocomposites because of its extraordinary properties such as high carrier mobility, large specific surface area, ultra high Young’s modulus, and excellent thermal and electrical conductivity [19–21]. The great engineering potential of this nanomaterial makes it applicable in the aviation industry, energy storage devices, microelectronics, memory chips, transparent and flexible devices, electromagnetic interference (EMI) shielding, and hydrogen storage systems [22–29]. Numerous studies have been carried out by using graphene as conductive filler to enhance the dielectric constant of polymers and very high dielectric constant can be achieved by using a very small quantity of graphene while retaining the flexibility, processability, and cost effectiveness of the polymers. The dielectric properties of graphene-based polymer composites can be optimized by controlling the dispersion state of graphene in polymer matrices and the interfacial interaction between them [21, 24]. As a precursor material, graphene oxide (GO) is the most reliable source for the preparation of graphene-based polymer composites. GO is a pseudo two-dimensional solid in bulk form which exhibits extraordinary optoelectronic [30], mechanical [31], and transport properties [32]. Other than the graphene scenario, GO has several standalone applications such as supercapacitors, memory devices, optoelectronics, photocatalysis, drug delivery, and composite materials [33–35]. The intriguing properties of GO have drawn the attention of the scientific community very recently. Therefore, it would be interesting to develop eco-friendly polymer/GO composite material suitable for commodity and technological applications.
Polyvinyl alcohol (PVA) is the most promising commodity polymer because of its applications in fuel cells, humidity sensors, food packaging, drug delivery, thin film transistors, optical waveguide sensors, and holographic gratings [36, 37]. PVA is also a potential material having high tensile strength and flexibility, high dielectric strength, good charge storage capacity, and excellent adhesive, emulsifying, and film-forming properties [38]. Another biopolymer, polyethylene glycol (PEG) is a water-soluble synthetic polymer widely used in pharmaceutical and cosmetic industries [39]. PEG has many attractive properties such as wide range of molecular weight, biocompatibility, low toxicity, and chain flexibility, and it has been used frequently in the production of polymer blends as it can improve the flexibility and ductility of rigid polymers. In the present study, PVA and PEG were chosen as the host polymers because both polymers are highly compatible with each other and form miscible blends through hydrogen bonding interactions between the hydroxyl groups of PVA and the ether linkage of PEG chains [40]. Both PVA and PEG are water soluble and can also form a variety of hydrogen bonding interactions with various fillers or additives.
In short, the aim of the present study is to fabricate GO reinforced polymer composites based on PVA and PEG using facile and eco-friendly colloidal processing method. In addition, the study also aims to explore the influence of GO content on the morphology and dielectric properties of PVA/PEG/GO composites. GO contains various reactive oxygen-containing functional groups on its surfaces and edges [21] and achieves molecular level dispersion in water-soluble polymers like PVA and PEG through strong hydrogen bonding interactions leading to enhanced composite properties. The structural, morphological, optical, and thermal properties of PVA/PEG/GO composites were investigated comprehensively. The dielectric constant and dielectric loss values of PVA/PEG/GO composites were investigated in order to check their feasibility as a flexible dielectric material for high-performance energy storage device applications such as embedded capacitors.
Experimental technique
Materials
Natural graphite powder (40 μm with purity >99.85 %) was supplied by Carbotech Engineers, Jaipur, India. Sulphuric acid (H2SO4, 98 %), potassium permanganate (KMnO4, 99.9 %), sodium nitrate (NaNO3, 99 %), and hydrogen peroxide (H2O2, 30 %) were purchased from S.D. Fine Chemicals, Mumbai, India, and used as received. PVA of molecular weight 85,000–124000 gm/mol (degree of hydrolysis 87–89 %) and high molecular weight PEG (100,000 gm/mol) was purchased from Sigma-Aldrich, India. Deionized water was used as a solvent for the preparation of PVA/PEG/GO composites. All the chemicals used in this study are of analytical reagent grade.
Synthesis of graphene oxide (GO)
In the present study, GO powder was synthesized from natural graphite powder using a modified Hummers method [41]. The detailed protocol for the synthesis of GO is given in our previous work [21, 24]. The synthesized GO powder was further used for the preparation of composites.
Synthesis of PVA/PEG/GO composites
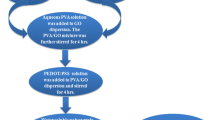
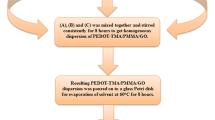
PVA/PEG/GO composites films with different GO loadings (0–3 wt%) were prepared using the facile colloidal processing technique. PVA powder was initially dissolved in deionized water at 75 °C for 4 h and later PEG powder was dissolved in deionized water at room temperature in a separate glass beaker. GO powder was also dispersed in deionized water in a separate beaker by ultrasonication process for 1 h. After that, the aqueous solutions of PVA, PEG, and GO were mixed together by vigorously stirring for 6 h until the uniform dispersion of GO in the polymer matrix is obtained. Finally, PVA/PEG/GO dispersion was casted onto a Teflon Petri dish and dried in a hot air oven at 60 °C for 5 h to get PVA/PEG/GO composite films. The resulting composite films were peeled off from the Teflon sheets and used for further study. The thickness of composite films was in the range of 70–80 μm. The feed compositions used for the preparation of PVA/PEG/GO composites are shown in Table 1. The step by step protocol for the synthesis of PVA/PEG/GO composites is given in Fig. S1.
Characterizations
FTIR spectra of PVA, PEG, GO, and PVA/PEG/GO composite films with different GO loadings were taken using Fourier Transform Infrared Spectrophotometer (Shimadzu, IRAffinity-1, Japan). The FTIR experiments were carried out in the wavenumber range 400–4000 cm−1 in a transmittance mode.
Raman spectroscopy of graphite, GO, and PVA/PEG/GO composite films was carried out using Raman Scattering Spectrometer (LABRAM HR 800) by using a 633-nm laser and a 1-μm spot size. For this, the samples were exposed for 10 s with a laser power of 10 mW at room temperature in backscattering mode. A charged coupled camera was used to collect the data in the wave number range 1000–3000 cm−1.
X-ray diffraction patterns of PVA/PEG/GO composite films at various GO loadings were obtained from Bruker AXS D8 focus advanced X-ray diffraction meter (Rigaku, Japan, Tokyo). The samples were scanned in the 2θ range from 5° to 80° using Cu-Kα radiation of wavelength λ = 1.54060 Å.
UV-vis absorption spectra of PVA/PEG/GO aqueous colloidal dispersions were obtained in the range of 190–600 nm with a Shimadzu UV-2401PC, UV-vis spectrophotometer.
Thermal stability of PVA/PEG/GO composite films was studied using Mettler Toledo, TGA/STDA851, thermogravimetric analyzer under N2 atmosphere. The samples were heated up to 450 °C at the rate of 10 °C/min for the measurements.
The dispersion state of PVA/PEG/GO composites films with different GO loadings was studied using crossed polarizing optical microscope (Olympus BX-53, Singapore) at a magnification of ×10.
The surface morphology of the PVA/PEG/GO composite films was evaluated by using scanning electron microscope (Carl Zeiss EVO/18SH, UK) and atomic force microscope (Nano Surf Easy Scan2, Switzerland). An accelerating voltage of 15 kV was applied to obtain the SEM images and the topographic atomic force microscopy (AFM) images were taken in tapping mode. For AFM study, the specimens were fixed on a glass slide using a piece of adhesive tape.
The dielectric properties of PVA/PEG/GO composite films were measured using Wayne Kerr 6500B (Chichester, West Sussex, UK) Precision Impedance Analyzer. The samples were placed inside a computer-controlled variable temperature oven, whose temperature can be programmed as per the need. In the present study, the dielectric properties were investigated in the wide range of frequencies from 50 Hz to 20 MHz and temperature in the range 40–150 °C with an accuracy of about +0.2 °C.
Results and discussion
FTIR spectroscopy studies
In the present investigation, FTIR spectroscopy was employed to explore the possible interaction between the functional groups of PVA, PEG, and GO which is schematically presented in Fig. S2. The FTIR spectra of PEG, PVA, and GO are shown in Fig. S3(a–c). The FTIR spectrum of pure PEG (Fig. S3(a)) shows characteristic bands at 2945 and 2879 cm−1 which are attributed to C-H stretching vibration and the range 1500–1300 cm−1 is attributed to C-H bending vibration. The FTIR bands in the range 1278–1091 cm−1 are attributed to stretching vibrations of alcoholic O-H and C-O-C ether linkage [42]. The band at 840 cm−1 is attributed to stretching vibration of C-C group. A characteristic band at 947 cm−1 which is attributed to C-OH group of PEG was also observed [43]. The FTIR spectrum of pure PVA exhibits several characteristics peaks as given in Fig. S3(b). The broad and strong absorption band in the range 3000–3400 cm−1 corresponds to the symmetrical stretching vibration of O-H group. The band appearing at 2928 cm−1 corresponds to C–H asymmetric stretching vibration of the alkyl group and the band at 1733 cm−1 is attributed to the C=O stretching vibration of vinyl acetate group of PVA [44, 45]. The band appearing at 1640 cm−1 can be assigned to the stretching vibration of C=O group and the band at 1438 cm−1 is attributed to CH2 bending [46]. The band at 1321 and 1243 cm−1 is respectively coming from the CH2 and C–H wagging vibrations. The band at 1084 cm−1 corresponds to the C–O stretching vibration of an acetyl group. The bands observed at 916 and 840 cm−1 are attributed to the skeletal vibration of PVA [37, 38]. The FTIR spectrum of GO is given in Fig. S3(c) from which the presence of different oxygen-bearing functional groups can be identified. The bands at 3441 and 2920 cm−1 are attributed to the O-H and C-H stretching vibration of GO, respectively. The band at 1730 cm−1 is attributed to C=O stretching vibration. The bands at 1396, 1255, and 1026 cm−1 are attributed to C-O stretching vibration of carboxylic, epoxy, and alkoxy groups of GO, respectively.
The FTIR spectra of PVA/PEG blend and PVA/PEG/GO composites are given in Fig. 1(a–g). Figure 1(a) depicts the FTIR spectrum of PVA/PEG blend film in which all the characteristic peaks of both PVA and PEG are seen. For PVA/PEG blend films, the band corresponding to C-H stretching vibrations were observed similar to the neat polymers. The peak at 1438 cm−1 corresponding to CH2 bending is shifted to 1427 cm−1 in PVA/PEG blend. Similarly, the peak at 1733 cm−1 corresponding to vinyl acetate group of PVA is also shifted to 1726 cm−1. These shifting of FTIR bands towards lower wavenumber suggest the occurrence of hydrogen bonding interaction between O-H groups of PVA and PEG polymer chains. Figure 1(b–g) shows the FTIR spectra of PVA/PEG/GO composites with different GO loadings. In the FTIR spectra of composites, the characteristics peaks of PVA, PEG, and GO were observed which indicates the successful preparation of PVA/PEG/GO composites.

FTIR spectra of PVA/PEG/GO composite films: (a) PVA/PEG blend, (b) 0.5 wt% GO, (c) 1 wt% GO, (d) 1.5 wt% GO, (e) 2 wt% GO, (f) 2.5 wt%, (g) 3 wt% GO
Raman spectroscopy studies
The Raman spectra of pristine graphite and GO are shown in Fig S4. The Raman spectrum of pristine graphite shows a strong G band at 1585 cm−1 which corresponds to the E2g phonon from sp2 carbon atoms and a weak D band at 1326 cm−1 which corresponds to sp3 carbon atoms and topological defects [21, 47]. In contrast, the Raman spectrum of GO displays a strong D band at 1365 cm−1 and a G band at 1634 cm−1 attributing to the size reduction of in-plane sp2 domains due to harsh chemical oxidation and ultrasonic exfoliation [24]. The intensity of D band is higher than that of G band and the intensity ratio of I D/I G is found to be around 0.83. The Raman spectra of PVA/PEG/GO composites with different GO loadings are depicted in Fig. 2. In the Raman spectra of composites the D band (1365 cm−1) is shifted to 1339, 1336, 1340, 1332, and 1348 cm−1 for 0.5, 1, 1.5, 2, and 3 wt% GO loadings, respectively. Similarly, G band (1634 cm−1) is shifted to 1598, 1592, 1594, 1601, and 1596 cm−1 for 0.5, 1, 1.5, 2, and 3 wt% GO loadings, respectively. Such shifts in the Raman bands of composites indicate significant interactions between PVA, PEG, and GO through hydrogen bonding corresponding to the homogeneous dispersion of GO in the polymer matrix.

Raman spectra of PVA/PEG/GO composite films: (a) 0.5 wt% GO, (b) 1 wt% GO, (c) 1.5 wt% GO, (d) 2 wt% GO, (e) 3 wt% GO
UV-visible spectroscopy studies
It is well understood that the homogeneous dispersion of GO in a polymer matrix is a very critical factor to achieve high-performance composites because most of the attractive properties of GO are associated with its individual sheets. Therefore, the prevention of aggregation and achieving molecular level dispersion of GO in the polymer matrix is very crucial while designing specific applications of graphene-based polymer composites. The UV-vis absorbance spectra of an aqueous solution of PVA, PEG, GO, and PVA/PEG blend are given in Fig. S5. The UV-vis absorbance spectrum of PVA showed a single characteristics absorption band at ∼200 nm which is associated with the residual acetate group of PVA [48]. The UV-vis absorbance spectrum of PEG aqueous solution showed an absorption band at ∼192 nm and the aqueous dispersion of GO showed a characteristic absorption band at ∼230 nm which is attributed to the π–π* transitions of the aromatic C=C bond [21, 24]. In addition, a shoulder at 310 nm was also observed in the UV-vis absorbance spectrum of GO which is attributed to n–π* transitions. The aqueous solution of PVA/PEG blend showed absorption band at ∼192 nm. UV-vis spectra of aqueous colloidal dispersion of PVA/PEG/GO composites are depicted in Fig. 3. It can be seen that the UV-vis absorbance spectra of PVA/PEG/GO, colloidal dispersion showed absorption band in the region 192–196 nm for different GO loadings in PVA/PEG blend matrix. In addition, a shoulder at 230 nm can be seen in all the composite samples indicating the presence of GO in the samples. The colloidal dispersion of PVA/PEG/GO composites was found to be homogeneous by visual inspection. The increasing the peak broadening by enveloping the GO in the composite suggest the presence of significant interaction between them which could be ascribed to the presence of the oxygen-containing functional groups on the surface of GO. Also, the hydrophilic characteristics of GO impart desired solubility and dispersibility in water, creating a stable GO suspension. Therefore, GO is compatible with many hydrophilic polymers such as PVA and PEG facilitating the formation of homogeneous composites.

UV-vis spectra of PVA/PEG/GO composites: (a) 0.5 wt% GO, (b) 1 wt% GO, (c) 1.5 wt% GO, (d) 2 wt% GO, (e) 2.5 wt% GO, (f) 3 wt% GO
X-ray diffraction studies
The XRD pattern of natural graphite powder, GO, PVA, and PEG is shown in Fig. S6. For natural graphite powder, a typical diffraction peak at 2θ = 26.42° with an interlayer distance of 0.33 nm can be observed, which corresponds to the (002) reflection plane. The XRD pattern of GO shows a characteristic peak at 2θ = 11.55° which corresponds to the interlayer distance of 0.82 nm [21, 24]. The interlayer distance has increased from 0.33 nm for graphite to 0.82 nm for GO. This indicates that the highly ordered crystalline structure of graphite is disrupted after oxidation. The increase in the interlayer distance is attributed to the intercalation and the bonding effect of oxygen-bearing functional groups revealing that graphite was oxidized effectively. The XRD pattern of pure PVA powder shows a diffraction peak at 2θ = 19.44° which is attributed to the existence of a high degree of hydrogen bonding interactions between hydroxyl groups [48–50]. The XRD pattern of pure PEG showed strong diffraction peak at 2θ = 19.34° corresponding to (120) crystal plane and weak diffraction peaks at 2θ = 23.33° which is attributed to concerted (112) and (032) crystalline reflection planes [51]. The XRD patterns of PVA/PEG blend and PVA/PEG/GO composite films are shown in Fig. 4(a–g). These patterns show two characteristic diffraction peaks at 2θ = 19.08° and 23.27° corresponding to PVA and PEG, respectively. Also, it can be seen that the diffraction peak of PVA (19.44°) and PEG (19.34°) are overlapped with each other in the XRD pattern of PVA/PEG blend and PVA/PEG/GO composite films. The XRD peak of GO was not observed in the XRD pattern of composite films which demonstrate that GO was fully exfoliated into individual graphene sheets and dispersed homogeneously in the polymer matrix.

XRD spectra of PVA/PEG/GO composites: (a) PVA/PEG blend, (b) 0.5 wt% GO, (c) 1 wt% GO, (d) 1.5 wt% GO, (e) 2 wt% GO, (f) 2.5 wt% GO, (g) 3 wt% GO
Thermal degradation characteristics
The TGA thermograms of PVA/PEG blend and PVA/PEG/GO composites are depicted in Fig. 5(a–g). The TGA thermograms of PVA/PEG blend film (Fig. 5(a)) show two-step decomposition. It has been reported that the incorporation of graphene-based materials into the polymer matrix can improve the thermal stability of polymer composites relative to the neat polymer [52]. The initial weight loss for PVA/PEG blend was observed from room temperature to 100 °C is attributed to the evaporation of water. The second decomposition step is observed in the 100–220 °C temperature range with minimum weight loss (∼20 %). In the third step of decomposition in the temperature range 220–450 °C, the blend sample showed maximum (∼98 %) weight loss. The TGA thermograms of PVA/PEG/GO composites with different GO loadings are depicted in Fig. 5(b–g) which showed decomposition in three stages. The initial weight loss stage occurs in the low-temperature range, i.e., room temperature to 100 °C is attributed to the evaporation of moisture or adsorbed water [53]. The second weight loss stage occurs in the temperature range 100–225 °C which could be due to the decomposition of different oxygen-bearing functional groups of GO. The third weight loss stage occurs in the temperature range 225–450 °C which could be due to splitting of polymer main chain. Thus, it can be seen that the thermal stability of PVA/PEG/GO composites films with different GO loadings has improved as compared to PVA/PEG blend. This could be due to the excellent dispersion of GO into the polymer matrix and the strong interfacial interaction between PVA, PEG, and GO through hydrogen bonding. The graphene oxide has a strong influence on the thermal degradation of PVA/PEG result in a charred layer which covers the surface of the polymer, thus interrupting the absorption of the oxygen required for the burning process and the attached polymer chains to the surface of the GO with resulting of molecules moving restriction under the temperature stimulus.

TGA thermograms of PVA/PEG/GO composites: (a) PVA/PEG blend, (b) 0.5 wt% GO, (c) 1 wt% GO, (d) 1.5 wt% GO, (e) 2 wt% GO, (f) 2.5 wt% GO, (g) 3 wt% GO
Morphological studies
The photographs of the as-synthesized PVA/PEG/GO composite films shown in Fig. 6a–f indicate the formation of the uniform dispersion of GO within the polymer matrix. From the photographs, it can be seen that the composite films have no cracks or holes and the films can be curled or folded, indicating the excellent flexibility of composites. To investigate the reinforcement effect and to understand the dispersion state of GO in the composites, optical microscopy was carried out and the results are depicted in Fig. S7. It is worthy to note that the dispersion of GO in the PVA/PEG blend matrix was fairly homogeneous without any discernible aggregation, indicating the good compatibility between GO and polymers. The dispersion state of GO in PVA/PEG blend matrix was further investigated by SEM analysis. Figure S8 shows microstructure of pure PVA film, PVA/PEG blend films, graphite, and GO. Pure PVA and PVA/PEG blend film display flat and smooth surface as shown in Fig. S8(a, b). SEM image of graphite displays flakes like microstructure, typical of highly crystalline natural graphite. SEM microstructures of PVA/PEG/GO composites with different GO loading are depicted in Fig. 7a–f. The SEM images of PVA/PEG/GO composites revealed a layered structure with uniformly dispersed GO sheets throughout the polymer matrix for all composite samples without any obvious aggregation. It is believed that the uniform dispersion of GO in the polymer matrix is caused by the compatibility between PVA, PEG, and GO and the strong interaction between them. The good dispersion of GO in PVA/PEG blend matrix is expected to result in an enhancement of dielectric constant of composites. AFM was employed in order to reveal the surface roughness of composite films and to obtain complementary data on the distribution of GO in PVA/PEG blend matrix. The AFM topographic images for PVA/PEG/GO composites in two and three dimensions with different GO loadings are illustrated in Fig. S9(a–g) and Fig. 8a–g, respectively. The surfaces of composite films were compared in terms of roughness parameters such as mean roughness (S a) and root mean square roughness (S q) and the data is depicted in Table 2. According to AFM results, the surface of the composite was found to be rough (S a = 73.63 nm for 3 wt% GO loading) while the surface of PVA/PEG blend film was relatively smooth (S a = 9.62 nm). In addition, the S q was also increased from 12.40 nm for PVA/PEG blend film to 88.32 nm for PVA/PEG/GO composite with 3 wt% GO loading. The increase in S q can be due to good compatibility between PVA, PEG, and GO and the homogeneous dispersion of GO into PVA/PEG blend matrix [54]. The good compatibility between PVA, PEG, and GO is attributed to the presence of numerous oxygen-containing functional groups on the surface of GO which forms strong hydrogen bonding interactions with polymer chains.

Photographs of PVA/PEG/GO composite films with different GO loading. a PVA/PEG (50/50) blend; b 0.5 wt% GO; c 1.5 wt% GO; d 3 wt % GO; e, f composite films are bent to demonstrate their flexibility

SEM micrographs of PVA/PEG/GO composites: a 0.5 wt% GO; b 1 wt% GO; c 1.5 wt% GO; d 2 wt% GO; e 2.5 wt% GO; f 3 wt% GO

3D AFM topographic images of PVA/PEG/GO composites: a PVA/PEG blend; b 0.5 wt% GO; c 1 wt% GO; d 1.5 wt% GO; e 2 wt% GO; f 2.5 wt% GO; g 3 wt% GO
Dielectric properties
In the present study, the dielectric properties of PVA/PEG/GO composites were investigated in order to check their feasibility for energy storage applications. Figure 9a–h shows variation in the dielectric constant of PVA, PVA/PEG blend, and PVA/PEG/GO composite films as a function of frequency (50 Hz–20 MHz) at various temperatures (40–150 °C). Table 3 summarizes the dielectric properties of the neat polymer, blend, and the composite films. The maximum value of dielectric constant obtained for pure PVA film (Fig. 9a) was 10.71 at 50 Hz and at 150 °C. Similarly for PVA/PEG blend film (Fig. 9b), the maximum value of dielectric constant was 31.22 at 50 Hz and at the same temperature 150 °C. Both PVA and PVA/PEG blend film showed a maximum value of dielectric constant at lower frequencies and with further increase in frequency the dielectric constant decreases.

a Dielectric constants of PVA film as a function of frequency at various temperatures. b Dielectric constants of PVA/PEG (50/50) blends as a function of frequency at various temperatures. c Dielectric constants of PVA/PEG/GO composites with 0.5 wt% GO loading as a function of frequency at various temperatures. d Dielectric constants of PVA/PEG/GO composites with 1 wt% GO loading as a function of frequency at various temperatures. e Dielectric constants of PVA/PEG/GO composites with 1.5 wt% GO loading as a function of frequency at various temperatures. f Dielectric constants of PVA/PEG/GO composites with 2 wt% GO loading as a function of frequency at various temperatures. g Dielectric constants of PVA/PEG/GO composites with 2.5 wt% GO loading as a function of frequency at various temperatures. h Dielectric constants of PVA/PEG/GO composites with 3 wt% GO loading as a function of frequency at various temperatures
For PVA/PEG/GO composite film with 0.5 wt% GO loading (Fig. 9c), the maximum value of dielectric constant was 60.11 at 50 Hz and at 150 °C and for composites with 3 wt% GO loading (Fig. 9h), the maximum value of dielectric constant was 644.39, at 50 Hz and at the same temperature. It is worthy to note that the dielectric constant of composites has increased with increase in GO loadings and all the high value of dielectric constants were obtained at lower frequencies. The GO as a kind of filler own high aspect ratio and have a functional groups to enhance dielectric constant effectively. The interfacial polarization, which normally exists between polymer and fillers, give rise to a much increased dielectric constant. This improvement of dielectric constant was due to Maxwell-Wagner-Sillar (MWS) effect in PVA/PEG/GO composites, thus making an important contribution to improving the dielectric constant at lower frequencies [55–57]. At lower frequencies, the rate of change of electric field is slow which provides sufficient time to permanent and induced dipoles to align themselves in the direction of the electric field leading to enhanced polarization. At higher frequencies, the dielectric constant show frequency independent behavior which indicates that the rotational motion of the polar molecules of composites is not rapid enough to attain the equilibrium with the electric field [21, 24]. Thus, the dielectric constant is low at high frequencies. The incorporation of GO into PVA/PEG blend matrix has resulted in the enhancement of dielectric constant which is 60 times higher than the dielectric constant of PVA and 20 times higher than the dielectric constant of PVA/PEG blend film. The effective dielectric constant of composites can be determined by the dielectric polarization and the relaxation mechanism of bulk composites. The high dielectric constant values of composites indicate their ability to store charges under the influence of electric field. Such composites can be useful in charge storage device applications such as embedded capacitors.
Dielectric loss (tan δ) is another important parameter of dielectric materials which results from the inability of the polarization process to follow the rate of change of applied electric field. In general, the dielectric loss results from distortional, dipolar, interfacial, and conduction loss in the dielectric materials [58, 59]. The variation in the dielectric loss of PVA, PVA/PEG blend, and PVA/PEG/GO composite films is depicted in Fig. 10a–h. The dielectric loss values of PVA, PVA/PEG blend, and PVA/PEG/GO composite films are high at low frequencies, and as the frequency increases, the dielectric loss decreases. This could be due to the interfacial polarization or Maxwell-Wagner-Sillar effect [60, 61]. According to MWS process, the presence of polymer-filler interface can lead to significant changes in the dielectric properties [62, 63]. The maximum value of dielectric loss for PVA (Fig. 10a) and PVA/PEG blend film (Fig. 10b) was found to be 1.91 and 3.04, respectively. For PVA/PEG/GO composite films with 0.5 wt% GO loading (Fig. 10c), the dielectric loss value is 3.66 and that of the composite with 3 wt% GO loading (Fig. 10h), the dielectric loss value is 5.58. The values of dielectric constant for all composites are much higher than the dielectric loss values. The enhancement in the dielectric constant values of composites is attributed to the homogeneous dispersion of GO into PVA/PEG blend matrix and the good compatibility between PVA, PEG, and GO. Thus, the dielectric properties of this type of composites can be tuned and optimized by controlling the dispersion state of GO in the polymer matrix and the interfacial interaction between GO and polymer.

a Dielectric loss tangents (tan δ) of PVA film as a function of frequency at various temperatures. b Dielectric loss tangents (tan δ) of PVA/PEG blend (50/50) film as a function of frequency at various temperatures. c Dielectric loss tangents (tan δ) of PVA/PEG/GO composites with 0.5 wt% GO loading as a function of frequency at various temperatures. d Dielectric loss tangents (tan δ) of PVA/PEG/GO composites with 1 wt% GO loading as a function of frequency at various temperatures. e Dielectric loss tangents (tan δ) of PVA/PEG/GO composites with 1.5 wt% GO loading as a function of frequency at various temperatures. f Dielectric loss tangents (tan δ) of PVA/PEG/GO composites with 2 wt% GO loading as a function of frequency at various temperatures. g Dielectric loss tangents (tan δ) of PVA/PEG/GO composites with 2.5 wt% GO loading as a function of frequency at various temperatures. h Dielectric loss tangents (tan δ) of PVA/PEG/GO composites with 3 wt% GO loading as a function of frequency at various temperatures
Conclusions
In the present study, smooth, uniform, and flexible PVA/PEG/GO composite films were obtained successfully using a simple colloidal processing technique. The structural studies of PVA/PEG/GO composites films by FTIR and Raman spectroscopy confirm the presence of GO in the composites. The PVA/PEG/GO composites films were investigated using optical microscopy which exhibits the homogeneous distribution of GO within the polymer matrix without any discernible agglomerations. The surface morphology confirmed the homogeneous dispersion of GO into PVA/PEG blend matrix. The thermal stability of composites has improved which could be due to strong hydrogen bonding interaction between functional groups of GO and polymer chains indicating good compatibility between PVA, PEG, and GO. The homogeneous dispersion of GO into PVA/PEG blend matrix and the strong interfacial interaction through hydrogen bonding between functional groups of GO and the polymeric chain has resulted in the enhancement of dielectric properties of composites. The dielectric constant for PVA and PVA/PEG (50/50) blend was found to be 10.71 and 31.22, respectively. The dielectric constant for PVA/PEG/GO composite with 3 wt% GO loading was found to be 644.39 which is 60 times greater than the dielectric constant of PVA and 20 times greater than the dielectric constant of PVA/PEG (50/50) blend film. Thus, we have successfully developed free standing and flexible PVA/PEG/GO composite films as a promising material with high dielectric constant and low dielectric loss for bio-friendly capacitors and other energy storage devices.
References
Wang Q, Zhu L (2011) J Polym Sci Part B: Polym Phys 49:1421–1429
Li Z, Fredin LA, Tewari P, Dibenedetto SA, Lanagan MT, Ratner MA, Marks TJ (2010) Chem Mater 22:5154–5164
Brochu P, Pei Q (2010) Macromol Rapid Commun 31:10–36
Li R, Xiong C, Kuang D, Dong L, Lei Y, Yao J, Jiang M, Li L (2008) Macromol Rapid Commun 29:1449–1454
Chu BJ, Zhou X, Ren K, Neese B, Lin MR, Wang Q, Baue F, Zhang QM (2006) Science 313:334–336
Arbatti M, Shan XB, Cheng ZY (2007) Adv Mater 19:1369–1372
Tortai JH, Bonifaci N, Denat A, Trassy C (2005) J Appl Phys 97:053304-1–053304-9
Zhou H, Cheng X, Rao L, Li T, Duan YY (2013) Acta Biomaterilia 9:6439–6449
Zhang QM, Li HF, Poh M, Xia F, Cheng ZY, Xu HS, Huang C (2002) Nature 419:284–287
Yao SH, Yuan JK, Dang ZM, Bai J (2010) Mater Lett 64:2682–2684
Huang XY, Xie LY, Jiang PK, Wang GL, Liu F (2009) J Phys D Appl Phys 42:245407
Huang XY, Xie LY, Hu ZW, Jiang PK (2011) IEEE Trans Dielect Elect Insul 18:375–383
Li J, Claude J, Norena-Franco LE, Seok SI, Wang Q (2008) Chem Mater 20:6304–6306
Li J, Seok SI, Chu B, Dogan F, Zhang Q, Wang Q (2009) Adv Mater 21:217–221
Xie L, Huang X, Wu C, Jiang P (2011) J Mater Chem 21:5897–5906
Li M, Huang X, Wu C, Xu H, Jiang P, Tanaka T (2012) J Mater Chem 22:23477–23484
Shen Y, Lin Y, Li M, Nan CW (2007) Adv Mater 19:1418–1422
Wu C, Huang X, Xie L, Wu X, Yu J, Jiang P (2011) J Mater Chem 21:17729–17736
Stankovich S, Dikin DA, Dommett GHB, Kohlhaas KM, Zimney EJ, Stach EA, Piner RD, Nguyen SBT, Ruoff RS (2006) Nature 442:282–286
He Q, Wu S, Gao S, Cao X, Yin Z, Li H, Chen P, Zhang H (2011) ACS Nano 5:5038–5044
Deshmukh K, Ahamed MB, Deshmukh RR, Pasha SKK, Sadasivuni KK, Ponnamma D, Chidambaram K (2016) Euro Polym J 76:14–27
Muhammed F, Syed K (2013) J Mater Sci Mater Electron 24:2202–2210
Parveen S, Veena C (2013) J Nanoparticles Res 14:1415–1422
Deshmukh K, Ahamed MB, Pasha SKK, Deshmukh RR, Bhagat PR (2015) RSC Adv 5:61933–61945
Park HD, Lee KY, Park SS, Lee SC, Kim HS, Kim NW (2013) Macromol Res 21:905–910
Wang Z, Nelson JK, Hillborg H, Zhao S, Schadler LS (2012) Adv Matter 24:3134–3137
Dang ZM, Yuan JK, Zha JW, Zhou T, Li ST, Hu GH (2012) Prog Mater Sci 57:660–723
Eda G, Fanchini G, Chhowalla M (2008) Nat Nanotechnol 3:270–274
Chan Y, Hill JM (2011) Nanotechnology 22:305403
Dikin DA, Stankovich S, Zimney EJ, Piner RD, Dommet GHB, Evmenenko G, Nguyen ST, Ruoff RS (2007) Nature 448:457–460
Gomez-Navarro C, Burghard M, Kern K (2008) Nano Lett 8:2045–2049
Dragoman D, Dragoman M (2007) Appl Phys Lett 91:203116–3
Khan U, May P, Neil AO, Coleman JN (2010) Carbon 48:4035–4041
Cheng Q, Tang J, Ma J, Zhang H, Shinya N, Qin LC (2011) Carbon 49:2917–2925
Krishnamoorthy K, Mohan R, Kim S (2011) J Appl Phys Lett 98:244101–244103
Prajapati GK, Gupta PN (2011) Physica B: Cond Mat 406:3108–3113
Pawde SM, Deshmukh K, Parab S (2008) J Appl Polym Sci 109:1328–1337
Pawde SM, Deshmukh K (2008) J Appl Polym Sci 109:3431–3437
Ramamohan K, Achari VBS, Sharma AK, Xiuyang L (2015) Ionics 21:1333–1340
Sawatari C, Kondo T (1999) Macromolecules 32:1949–1955
Hummers WS, Offeman RE (1958) J Am Chem Soc 80:1339
Ahmad MB, Tay MY, Shameli K, Hussein MZ, Lim JY (2011) Int J Mol Sci 12:4872–4884
Fan W, He M, Wu X, Chen B, Hu B (2015) J Chromatrography A 1418:36–44
Ahmad J, Deshmukh K, Hagg MB (2013) Int J Polym Anal Charac 18:287–296
Ahmad J, Deshmukh K, Habib M, Hägg MB (2014) Arab J Sci Tech 39:6805–6814
Deshmukh K, Ahmad J, Hagg MB (2014) Ionics 20:957–967
Ferrari AC, Robertson J (2001) J Phys Rev B 64:075414–075513
Deshmukh K, Ahamed MB, Deshmukh RR, Bhagat PR, Pasha SKK, Bhagat A, Shirbhate R, Telare F, Lakhani C (2016) Polym Plast Technol Eng 55:231–241
Morariu S, Rusu D (2013) Soft Matter 9:1244–1253
Qian XF, Yin J, Huang JC, Yang YF, Guo XX, Zhu ZK (2001) Mater Chem Phys 68:95–97
Zhu S, Chen J, Li H, Cap Y (2013) Appl Surf Sci 264:500–506
Potts JR, Dreyer DR, Bielawski CW, Ruoff RS (2011) Polymer 52:5–25
Zhang J, Qiao J, Jiang G, Liu L, Liu Y (2013) J Power Sources 240:359–367
Mallakpour S, Abdolmaleki A, Khalesi Z, Borandeh S (2015) Polymer 81:140–150
Pawde SM, Deshmukh K (2008) J Appl Polym Sci 110:2569–2578
Pasha SKK, Deshmukh K, Ahamed MB, Chidambaram K, Mohanapriya MK, Arunai Nambi Raj N, Adv. Polym. Tech. (2015) doi:10.1002/adv.21616
Mohanapriya MK, Deshmukh K, Ahamed MB, Chidambaram K, Pasha SKK (2015) Int J Chem Tech Res 8:32–41
Mohanapriya MK, Deshmukh K, Ahamed MB, Chidambaram K, Pasha SKK (2016) Mater Today Proceed 3:1864–1873
Mohanapriya MK, Deshmukh K, Ahamed MB, Chidambaram K, Pasha SKK (2016) Adv. Mater. Lett. doi:10.5185/amlett.2016.6555
Deshmukh K, Ahamed MB, Polu AR, Sadasivuni KK, Pasha SKK, Ponnamma D, AlMaadeed MAA, Deshmukh RR, Chidambaram K (2016) J. Mater. Sci: Mater. Electron. doi:10.1007/s10854-016-5267-x
Deshmukh K, Ahamed MB, Deshmukh RR, Pasha SKK, Chidambaram K, Sadasivuni KK, Ponnamma D, AlMaadeed MAA (2016) Polym. Plast. Tech. Eng. doi:10.1080/03602559.2015.1132451
Sadasivuni KK, Ponnamma D, Kumar B, Strankowski M, Cardinaels R, Moldenaers P, Thomas S, Grohens Y (2014) Comp Sci Tech 104:18–25
Sadasivuni KK, Castro M, Saiter A, Delberilh I, Feller JF, Thomas S, Grohens Y (2013) Mater Lett 96:109–112
Acknowledgments
One of the authors, Kalim Deshmukh, would like to extend his kind gratitude towards the management of B. S. Abdur Rahman University, Chennai-600048, TN, India, for providing Junior Research Fellowship (JRF) to carry out this research work.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 2171 kb)
Rights and permissions
About this article

Cite this article
Deshmukh, K., Ahamed, M.B., Sadasivuni, K.K. et al. Graphene oxide reinforced polyvinyl alcohol/polyethylene glycol blend composites as high-performance dielectric material. J Polym Res 23, 159 (2016). https://doi.org/10.1007/s10965-016-1056-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-016-1056-8