
Abstract
The isotactic polypropylene – modified silica (iPP/SiO2) hybrid nanocomposites, obtained by grafting polypropylene chains on amine-functionalized silica particles during reactive blending, were studied. It was found that use of amine-functionalized silica and PP-g-MA as a compatibilizer improved dispersion of nanoparticles. Nanosilica, especially surface-modified, revealed some nucleation activity towards iPP, manifesting in an increase of the crystallization temperature and reduction of spherulite size. iPP/silica nanocomposites exhibit highly improved thermo-oxidative stability, due to formation of a silica protective layer, limiting the polymer volatilization rate. Nanocomposites demonstrate enhanced stiffness and strength, but at higher silica content the ductility is nearly lost due to presence of big agglomerates, acting as critical-sized structural flaws. At low silica concentrations dispersion was improved and big agglomerates were not observed. Consequently, iPP/PP-g-MA/am-SiO2 nanocomposites with low silica content demonstrate high ductility and enhanced impact resistance, related to reinforcing effect of well dispersed silica particles.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polymeric materials generally have moduli and strength lower than metals or ceramics. Their mechanical and some other properties can be effectively improved at a relatively low cost by reinforcing with micro- or nanosized stiff inorganic filler particles [1, 2]. Using such an approach, the modulus and strength of a polymer can be efficiently enhanced while maintaining its high ductility, low density and easy processing. This can be achieved in some cases even at relatively low nanofiller content. In fact, it has been demonstrated that addition of a small amount of selected nanofillers to polymers can improve their mechanical properties [3–6], barrier properties [7], thermal stability and chemical resistance [8]. Moreover, the drawbacks typically associated to the addition of traditional microfillers, such as embrittlement and loss of optical clarity, can be generally avoided [3]. The primary reason for adding inorganic nanofillers to polymers is to improve their mechanical performance [5]. One of the major requirements is then reaching the balance between the stiffness/strength and the toughness as much as possible [9].
The nanofillers used for fabrication of nanocomposites may have quasi-spherical (e.g. metals, metal oxides, minerals), platelet (clays), or fibrous (nanofibers or nanotubes) shapes. Among spherical particles, SiO2 (silica) appears especially important due to low cost, variety of available particle sizes and facility of surface modification. Therefore, polymer-silica nanocomposites have attracted substantial academic and industrial interest and actually are the most commonly inorganic-organic nanocomposites reported [5]. Numerous grades of silica nanoparticles are available commercially, usually in the form of powder or colloid. Nanosilica powder is produced in vast quantities mainly by the fuming method [10]. Fumed silica is a fine amorphous powder, considered non-toxic and non-irritating. It has an extremely large surface area (50–300 m2/g) and smooth nonporous surface of fine particles, which could promote good physical contact between the filler and the polymer matrix [11]. Surface of particles contains Si–O–Si units and Si–OH bonds which facilitate easy modification trough chemical functionalization. The primary nanoparticles of fumed silica, usually about 10–20 nm in diameter, tend to physically fuse together to form aggregates. These aggregates form bigger agglomerates that can be destroyed, at least partially, during the melt compounding process with a polymer.
Due to a common tendency of nanoparticles to agglomerate they are difficult to be uniformly dispersed in polymers by conventional mixing techniques. In the case of bare, unmodified silica aggregation and agglomeration tendencies are very strong due to hydrogen bonds formed by silanol groups existing on surface of particles. These bonds hold individual silica particles together and agglomerates remain intact even under the best mixing conditions, if stronger filler-polymer interaction is not present [12]. Such agglomerates can act upon loading as structural flaws that induce a premature fracture of the material. Therefore, the main challenge is to eliminate or at least significantly reduce aggregation and agglomeration to obtain a fine dispersion of nanosized primary silica particles. In conventional compounding a good dispersion may be achieved only by a suitable surface chemical or physical modification of nanoparticles [13].
Because of combination of low cost, high chemical resistance, and well balanced mechanical properties, isotactic polypropylene (iPP) is one of the most widely used thermoplastics. Still, for some applications its properties, as e.g. the low-temperature impact resistance or the long-term creep need to be improved. Nowadays, many reports have been focused on addition of nanosilica to enhance mechanical properties of polyolefins, including iPP [4, 5]. It was demonstrated that introduction of a small amount of silica nanoparticles (usually in range of 0.5–6 %) into iPP matrix may lead to substantial improvement of the mechanical properties of the resulting composite [4, 5, 14–28]. A remarkable progress was achieved through surface functionalization of nanofillers as well as by introduction of compatibilizers. For non-polar polymers, such as iPP, compatibilizers should be used in addition to filler modification. PP grafted with maleic anhydride or acrylic acid (PP-g-MA and PP-g-AA, respectively) are the most common compatibilizers used for iPP [29].
In the present work we focused on preparation and characterization of morphology, thermal and mechanical performance of systems obtained by reactive blending of plain iPP, PP grafted with maleic anhydride (PP-g-MA) and SiO2 nanoparticles, surface functionalized with amine groups, taking advantage of the high efficiency of amine-anhydride reaction between amine groups at the surface of the silica nanoparticle and MA groups grafted to PP backbone. The modified silica particles were expected to disperse in iPP/PP-g-MA matrix much easier than bare silica exhibiting a strong agglomeration tendency [30]. This could eventually result in obtaining the balanced PP/silica nanocomposite by simple blending. The reactivity of molten PP-g-MA with model amine species has been recently assessed, showing quantitative conversion to imide [31]. Such an approach of one-step reactive blending involving amine-anhydride reaction was applied successfully to obtain polypropylene-POSS organic-inorganic hybrids [32–34]. The influence of grafting of PP-g-MA on amine-functionalized silica on the morphological characteristics and properties of obtained nanocomposites was studied in this work. In addition to reactive blends, physical blends of plain iPP homopolymer with the unmodified silica and ternary compositions of iPP, PP-g-MA and unmodified silica, were examined for comparison.
Experimental
Materials
Isotactic polypropylene, iPP (Malen-P, F-401, Basell Orlen Polyolefins; Mw = 297,000, Mw/Mn = 5.27, MFR (2.16 kg, 230 °C) = 3 g/10 min, density 0.904 g/cm3) was used as a matrix polymer. The maleic anhydride grafted polypropylene, PP-g-MA (Polybond 3150, Chemtura, USA; 0.5 wt.% of MA, Mw ~ 330 000, MFR = 50 g/10 min, density 0.91 g/cm3) was used as a reactive compatibilizer.
Two grades of hydrophilic fumed silica were used in this study: Aerosil 200 (density 2.28 g/cm3, BET specific surface area of 200 ± 25 m2/g, average primary particle size 12 nm) and Aerosil OX50 (BET specific surface area of 50 ± 15 m2/g, average primary particle size 40 nm), both obtained from Evonik Industries (Germany). They were used to prepare nanocomposites either as-received or surface modified with amine groups (hereafter coded S200, S50 and am-S200, am-S50, respectively).
(3-Aminopropyl)triethoxysilane, APTES (Sigma-Aldrich; M = 221.37 g/mol, ρ = 0.946 g/ml) was used as received. Toluene (Chempur) was dried over 4 Å molecular sieve and distilled prior to use.
Functionalization of nanoparticles
Introduction of amine groups onto surface of silica nanoparticle was done according the following procedure: 4 g of SiO2 nanoparticles (dried 12 h at 100 °C in vacuum) was dispersed in 100 ml of dry toluene under protection of flowing dry nitrogen. After ultrasonication for 120 min at room temperature, a solution of APTES (10 ml) in dry toluene (50 ml) was added drop-wise to the reaction flask over 25 min, still under nitrogen protection. The mixture was then kept for 240 min at 80 °C in ultrasonic environment under dry nitrogen protection. Next the reaction flask was transferred into an oil bath heated up to 110 °C and refluxed for 180 min under nitrogen. After cooling to room temperature, the mixture was filtered and the collected APTES-modified nanoparticles were washed several times with toluene in order to remove the residual unreacted APTES. Finally, the particles were dried under vacuum at 60 °C for 36 h, weighed and stored for future use.
Sample preparation
Prior to blending the silica nanofillers and PP-g-MA were dried at T = 100 °C for 12 h. iPP was used as received.
The binary iPP/SiO2 and ternary iPP/PP-g-MA/SiO2 and iPP/PP-g-MA/am-SiO2 nanocomposites were prepared by melt compounding, using the PlastiCorder (Brabender, Germany) internal mixer for 10 min at the temperature of 180 °C and the rotor speed of 60 rpm. In the first step iPP and PP-g-MA were loaded to the mixer in the desired proportion. After melting and approx. 1 min. mixing the nanofiller was added slowly to the molten blend and mixing continued for the next 9 min. Upon blending of iPP and PP-g-MA with SiO2 or am-SiO2 the anhydride reaction with either silanol or amine groups at the surface of filler particles was likely to take place, therefore that blending can be considered as the reactive blending process. The compositions prepared are summarized in Table 1.
The 0.5 mm or 1 mm thick sheets and 4 mm thick plates were produced from prepared compositions by compression molding at 180 °C in a laboratory press under the pressure of 50 bar. After molding the sheets and plates were fast cooled down to room temperature.
Characterization
NMR
29Si single pulse (SP MAS) and 29Si-1 H cross polarization (CP MAS) solid state magic angle spinning NMR spectra were recorded on the Bruker Avance III 400 MHz instrument operating at 294 K. Solid materials were carefully compacted into a rotor (zirconium oxide, 4 mm in diameter) and spun at 8 kHz. 29Si chemical shifts were calculated relative to an external standard 2, 2-dimethyl-2-silapentane-5-sulfonate (DSS).
DSC
Thermal analysis of compression molded samples was carried out with a differential scanning calorimeter (TA 2920, Thermal Analysis, USA). The thermograms were recorded at the heating/cooling rate of 10°/min, under nitrogen flow (20 ml/min). The overall crystallinity was estimated on the basis of the enthalpy of fusion. The value ∆Hf = 209 J/g was assumed for calculations [35].
TGA
Thermogravimetric measurements were performed with TGA Hi-Res 2950 instrument (Thermal Analysis). Samples were heated from room temperature up to 800 °C at the rate of 20 °C/min in the atmosphere of nitrogen or air (flow rate 60 ml/min). The degradation temperature Td was determined at the temperature corresponding to the maximum rate of the mass loss.
SEM
The morphology of nanocomposites was examined with a scanning electron microscope (JSM 6510 LV, Jeol Co, Japan) operating in the high vacuum mode and accelerating voltage of 15 kV. The compression molded samples were cryo-fractured. To prevent charging the samples were coated with thin layer of gold (ca.20 nm) by ion-sputtering (Jeol JFC-1200) prior to examination.
TEM
Details of morphology and nanofiller dispersion were examined with a transmission electron microscope (BS 500, Tesla, Czech Republic), operating at 90 kV. Samples, in the form of ultra-thin sections approx. 60 nm thick, were prepared by cryo- ultrasectioning with an ultramicrotome (PowerTome PC, Boeckeler, USA) equipped with a 35o diamond knife (Diatome, Switzerland). No staining or any other chemical treatment was applied to sections prior to observation.
From TEM micrographs the silica dispersion was evaluated using the image analysis software ImageJ (National Institutes of Health, USA). Size determinations were made by evaluating at least 500 agglomerates for each sample.
SALS
The samples in the form of films, approx. 30 μm thick, were examined with small angle light scattering technique to check a spherulitic morphology. A He-Ne lase (λ = 632.8 nm) was used to generate the Hv scattering patterns.
WAXS
The crystalline structure of nanocomposites was probed with X-Ray diffraction. The diffraction data were collected in the reflection mode with the DRON diffractometer (Russia), using CuKα radiation (λ = 0.154 nm). The 2Θ scans were collected with the step of 0.05°. From obtained diffractograms the phase composition was evaluated, including the crystallinity degree and the content of the β-phase. For accurate determination of phase structure the peak separation procedure was applied, using the fitting program FITYK [36].
Estimation of the β contribution to the crystalline phase, Kβ based on the Turner-Jones equation [37]:
where Iα(110), Iα (040) and Iα (130) are the integral intensities of the strongest peaks of α-form attributed to the (110), (040) and (130) planes of the α monoclinic cell, respectively, while Iβ(110) is the intensity of the strongest (110) diffraction peak of the trigonal β-form, located at 2Θ = 16.2°.
The lateral size of crystallites was evaluated from the half-width of the (110) and (040) diffraction peaks of α-PP using the Scherrer formula [38]:
where L hkl is a crystallite dimension in the direction perpendicular to the (hkl) plane, λ is the X-ray wavelength, β is the half-width of a diffraction peak, corrected for instrumental broadening, and Θ is the Braggs diffraction angle.
SAXS
The lamellar structure of samples was probed with 2-dimensional small angle X-ray scattering (2-D SAXS). The Kiessig-type camera with sample detector distance of 1.2 m was coupled to a X-ray CuKα low divergence microsource, operating at 50 kV and 1 mA (sealed-tube micro-focus source integrated with multilayer collimation optics, producing a highly collimated beam with a divergence of 0.8 × 0.8 mrad2; GeniX CU-Low Divergence by Xenocs, France). The collimation optics was combined with 2 additional hybrid scatterless slits systems forming the beam (Xenocs) placed between the multilayer optics and the sample stage. The two slit systems were separated by 1200 mm. The scattering produced by the sample was recorded with the Pilatus 100 K solid state area detector of the resolution of 172 × 172 μm2 (Dectris, Switzerland). The long period (LP) was determined from one-dimensional sections (background and Lorentz corrected) of 2-D patterns using the Bragg’s law.
Mechanical properties
The tensile tests were carried out using the Model 5580 Testing Machine (Instron Corp., USA) at room temperature. Specimens of the gauge length of 25 mm and thickness of 1 mm (specimen 1BA according to ISO 527-2) were cut out from compression molded sheets. The deformation rate was 5 mm/min.
The impact properties were evaluated by notched Izod impact test at room temperature, according to the ISO 180/1 A standard. The instrumented impact tester equipped with a 4 J hammer (Resil 5,5; CEAST S.p.A., Italy) was used. Additionally, the tensile impact test was performed according to the ISO 8256 standard with the same apparatus (v = 2.6 m/s, E = 1 J) and using unnotched specimens of the gauge length of 15 mm, width of 10 mm and thickness of 0.5 mm.
In every tensile or impact test at least 5 specimens of each composition was tested.
DMA
The dynamic mechanical properties of the deformed samples were measured with the TA Q800 dynamic mechanical thermal analyzer (Thermal Analysis, USA) in the single-cantilever bending mode. The specimens, in the form of 40 mm long, 10 mm wide, and 1 mm thick bars, were cut out from compression molded sheets. The storage modulus (E’), loss modulus (E”) and loss factor (tan δ) were determined at a constant frequency of 1 Hz as a function of temperature, which varied from –60 °C to 150 °C at a heating rate of 2 °C/min.
Results and discussion
Particle modification and grafting
A good dispersion of nanosilica particles can be obtained by suitable surface chemical or physical modification [13]. In the present study we intended to take advantage of amine-anhydride reaction for grafting of maleated polypropylene onto amine-functionalized surface of SiO2 particles. In order to achieve that the silica particles were treated with aminoorganosilane (APTES) to obtain a surface layer containing top –NH2 functional groups that would allow further grafting of PP-g-MA chains.
Silanization is a common approach to enhance the compatibility between polar inorganic nanoparticles and a hydrophobic polymer matrix [5]. Commonly used tri-functional alkoxysilanes, as APTES, spontaneously self-condensate in the presence of even trace of water (which is necessary for initiation of the APTES-silica reaction) and form cross-linked silane multilayers of complex structure around silica particles. Efficient top-surface functionalization with aminosilanes has been reported [39–41] on flat surfaces when the presence of water in the system was strictly controlled to permit the formation of a surface silane monolayer instead of complex multilayered coating. The density of such a monolayer was reported as 5.3 molecules/nm2 and the thickness as 0.7 nm. However, similar surface functionalization of nanoparticles in suspension is much more complicated than flat model substrates due to the potential particle aggregation, Brownian motion, solvent effects etc. Particle aggregation phenomena and interparticle cross-linking through siloxane bridges may appear as a result of uncontrolled reaction conditions. In the case of nanoparticles dispersed in aqueous suspension, aminosilanization can be even more complex process due to the various possible reactions of aminosilanes with water. It was observed [42–44] that aminosilane treatment of nanoparticles in a water-rich suspension produces cross-linked silane multilayers with amine groups mostly internalized or sequestered. Choi et al. [45] described the complex 3D network of APTES containing also cyclic structures resulting from hydrogen bonding of primary amines and hydroxyl groups. Both cyclic structured amines and sterically hindered amines trapped inside the silane coating are inaccessible to subsequent reactions, as e.g. with MA groups. The only amine groups accessible are those located on the top of the silane layer. In order to obtain such structure the amount of water in the system must be controlled very carefully [42].
In the procedure adopted in this study aminosilanization of nanoparticles with APTES was carried out in a dispersion in dry toluene. The amount of water present was minimized to obtain particles with thin coating and significant fraction of surface –NH2 groups that are able to interact later with the target molecular species. APTES is soluble but do not hydrolyze or self- condense in toluene hence APTES molecules remain unreacted in the dry toluene solution. They can hydrolyze and subsequently condense only when a small amount of water is let in the system in the form of the water surface layer on the nanoparticles introduced to toluene. This water is chemi- and physisorbed to the hydrophilic particles and does not diffuse into the toluene due to its hydrophobic nature. Therefore, it initiates condensation of APTES only on the surface of nanoparticles.
The silane content in modified particles was assessed with TGA. Unfortunately, TGA is unable to differentiate between covalently bonded organic coating and non-reacted, physicosorbed compounds. Further, the silane grafting density estimated from silane content obtained by TGA can differ from the accessible amine-groups on the top surface of an APTES layer, since an unknown number of amines will be internalized within silane coating, making them unavailable to subsequent reaction with MA. In order to reduce an amount of non-reacted adsorbed compound the particles were washed several times with toluene prior to TGA analysis. From obtained TGA thermograms of pristine and modified particles the net weight loss related to APTES volatization was determined and then the amount of the silane coating, layer thickness and silane grafting density were estimated. The pristine SiO2 is thermally stable at temperature higher than 700 °C and the weight loss is below 0.5 %, which is attributed to the dehydration of Si-OH and the water adsorbed on the surface of SiO2. The weight loss of modified particles is related mainly to thermal decomposition of APTES. The non-volatile part of APTES was assumed to be converted into silica (SiO2). Due to the stoichiometric relationship of SiO2 (1:2), the remaining third oxygen in APTES was considered to be volatile (in the form of H2O), which sets the molecular weight of the volatile fraction of APTES to 74 g/mol [46].
The grafting ratio Gr (silane-to-silica weight ratio), silane layer thickness, d, and silane grafting density, ρs, were calculated using the following equations:
Where Wam-SiO2 SiO2 is the difference of the weight loss of silanized and pristine samples, M = 221.4 g/mol and Mvt = 74 g/mol are the molecular mass of APTES and its volatile part, respectively, S is the specific surface area of the silica (2x1020 and 0.5x1020 nm2/g for S200 and S50 silica, respectively), and NA the Avogadro number. The results of evaluation are given in Table 2.
The results show that for both grades of silica used in this work the amount of APTES bonded to the silica surface is about 15 wt.%. However, because of different particle size and hence the specific surface area the estimated average layer thickness layer is approx. 0.8 nm for am-S200 and 3.3 nm for am-S50, which suggests a silane monolayer formed in am-S200 and a multilayer coating in am-S50. In the case of monolayer practically all amine groups are likely on the top of the layer and should be accessible for subsequent reaction with MA groups. In the case of multilayer structure in am-S50 some part of –NH2 groups is probably trapped inside the silane multilayer and become inaccessible.
To find out whether particles of am-SiO2 really have accessible amine groups on the top surface the NMR studies were carried out. Solid state NMR is a powerful tool for elucidating the molecular structure of the materials such as particles studied here. Figure 1 shows the 29Si single pulse (SP) and 29Si-1 H cross polarization (CP) magic angle spinning (MAS) spectra of the pristine S50 and aminosilanized am-S50 samples. The pristine, non-aminosilanized silica nanoparticles exhibited peaks in the region from −90 to −120 ppm that indicate the presence of only Qn (Si) species in both SP and CP mode (cf. Fig. 1a). Peak intensities in the SP mode can suggest that relative concentrations follow the trend Q4 > Q3 > Q2 Si-species, although the intensity of the Q3 (Si) resonance was observed to be the highest in CP mode. This may be expected as Q3and Q2 are capable of cross-polarization transfer from neighboring protons whereas Q4 is not. However, this observation also suggests that the fraction of Q3 species is much higher than Q2 species, particularly as the density of neighboring protons in Q2 is twice that in Q3.

29Si single pulse (SP) and 29Si-1 H cross polarization (CP) magic angle spinning (MAS) spectra of the pristine S50 (a), and aminosilanized am-S50 (b) nanoparticles
Sen and Bruce [42] demonstrated that material obtained from condensing/polymerizing APTS in water (no silica particles added) does not exhibit any peak in the region of −90 to −120 ppm indicating the absence of Qn (Si) species. Instead, the peaks related to Tn(Si) species were observed in the range of −50 to −70 ppm, as a consequence of the presence of the Si bonded to propyl amine group. Three different Tn (Si) sites were observed with relative concentrations of T3 > T2 > T1 (Si) in the SP mode, but only two types of T sites (T2, T3) in the CP mode [42]. In the case of silica particles aminosilanized in presence of water the Tn as well Qn resonances were observed [42], related to aminosilane coating and silica core, respectively. The species containing amine groups present in the system can be T1, T2 or T3 (Si) types as a consequence of the uncontrolled surface condensation of aminosilane in water environment [42, 47]. In fact, comparable intensities of T2 and T3 resonances were reported for silica particles silanized in water [42].
The am-S50 nanoparticles aminosilanized with reduced presence of water also exhibited in NMR spectra two types of Si environments, T and Q (Fig. 1b). SP mode intensities of Si resonances in T sites are low, however, become higher in cross-polarization mode owing spin population energy transfer from neighboring proton nuclei. In both SP and CP modes only T2 and T3(Si) sites were detected indicating two types of T(Si) environments, yet T3(Si) sites produce significantly higher signal than T2, in contrast to particles modified by silanization in aqueous environment. That difference in T2 and T3(Si) intensities suggests that larger fraction of NH2 groups is located at the top surface of silane layer coating a particle than those trapped inside the silane multilayer. These surface amine groups should be easily accessible to further reaction with MA upon compounding. An additional observation is that intensities of Q2(Si) species were significantly reduced in amino-functionalized material; most likely due to the loss of Si-OH groups by conjugation with aminosilane through dehydration.
The NMR results indicate a significant fraction of amine group in am-S50 located on the top surface of particles, thus being easily accessible for further reactions upon compounding. This allows to consider the obtained am-SiO2 particles as properly surface-functionalized.
The obtained amine-functionalized silica particles were introduced to the mixture of molten iPP and PP-g-MA in the process of melt-compounding. The reaction between MA and -NH2 groups on the surface of silica nanoparticles and consequently some coating of these particles with PP chains was expected to occur during that blending, according to the scheme presented in Fig. 2. The reaction should actually lead to formation of the organic-inorganic hybrids (PP-silica) in the nanocomposite.

The scheme of the melt grafting of PP-g-MA onto amine-functionalized SiO2 nanoparticle. Only one aminoorganosilane chain instead of silane layer is shown for clarity
The occurrence of similar grafting reaction between anhydride groups of PP-g-MA chains and surface amine groups of POSS particles, and in this way formation of PP-POSS hybrids, was evidenced by FTiR for reactive blending of PP-g-MA with amine functionalized POSS [34] - the carboxylic band at 1710 cm−1 of MA was replaced by a stronger peak at 1703 cm−1 characteristic of the imide group. Since the POSS cage molecule can be considered as the smallest silica particle, one can expect that the analogous reaction between surface amine groups and PP-g-MA can occur also on the surface of functionalized nanosilica particles during melt compounding. Unfortunately, FTiR analysis of the reactive composite of iPP, PP-g-MA and am-SiO2 obtained in this study did not detect development of such imide band, probably because of too low concentration of reacting species in the composite.
Dispersion of nanoparticles
The structure and morphology of composites obtained by blending was examined by SEM and TEM microscopy, light scattering (SALS), and X-Ray scattering (WAXS, SAXS) measurements. Melting temperature of crystallites and the overall crystallinity of composites were assessed by DSC.
Preliminary SEM observations (cf. Fig. 3) revealed generally uniform dispersion of silica particles, yet with a tendency to form relatively small, loose aggregates. In iPP/SiO2 and iPP/PP-g-MA/SiO2 composites with unmodified silica (of both S200 and S50 grades) the bigger and more dense agglomerates, usually a few micrometers in diameter were also found, cf. Fig. 3a, b. On the contrary, such big agglomerates were observed only occasionally in the iPP/PP-g-MA/am-SiO2 nanocomposites.

Exemplary SEM micrographs of the freeze-fracture surface of the PP-SiO2 nanocomposites, each containing 5 wt.% of S200 silica: a iPP/S200; b iPP/PP-g-MA/S200; c iPP/PP-g-MA/am-S200. The scale bar represents 5 μm
Transmission electron microscopy (TEM) images provide direct inspection of the dispersion of silica nanoparticles in the PP matrix, more detailed than SEM. Figures 4 and 5 present exemplary TEM micrographs of ultra-thin sections of nanocomposites containing 5 wt.% of S200 and S50 silica, respectively. Low magnification micrographs of all studied composites (cf. Figs. 4a–c and 5a–c) confirm previous SEM observations that the silica particles tend to form both aggregates and large, dense agglomerates. These aggregates and agglomerates are dispersed quite uniformly in the polymer matrix. The tendency to form agglomerates is especially the case of iPP/S200 (Fig. 4a) and iPP/PP-g-MA/S200 (Fig. 4b) nanocomposites containing untreated S200 silica of smaller primary particle size (<d > =12 nm), in which dense agglomerates exceeding 2 μm in diameter were frequently observed. Examination of samples with various silica content indicate that in general, an amount and size of agglomerates increases with increasing filler percentage (only the micrographs of nanocomposites containing 5 wt.% of SiO2 are presented for brevity). TEM micrographs taken at higher magnification (Figs. 4d–f and 5d–f) reveal that the size of agglomerates in nanocomposites studied depends on both the size of primary particles and their surface treatment as well as the presence of PP-g-MA compatibilizer.

TEM micrographs of iPP/S200 (a, d); iPP/PP-g-MA/S200 (b, e) and iPP/PP-g-MA/am-S200 (c, f) nanocomposites, each containing 5 wt.% of S200 silica. Insets in figures (a)–(c) show agglomerate size distributions determined by image analysis

TEM micrographs of iPP/S50 (a, d); iPP/PP-g-MA/S50 (b, e); iPP/PP-g-MA/am-S50 (c, f) nanocomposites, each containing 5 wt.% of S50 silica. Insets in figures (a)–(c) show agglomerate size distributions determined by image analysis
In the case of pristine S200 particles dispersed in plain iPP the particles are arranged mostly in agglomerates, approx. 200-1000 nm in size. Moreover, these agglomerates are frequently interconnected to form even larger and more developed structures (Fig. 4a, d). When the same untreated silica is dispersed in iPP with 2.5 wt.% of PP-g-MA as compatibilizer the agglomerates become smaller, not exceeding 500 nm, although some big agglomerates still can be found (Fig. 4b, e). In the nanocomposite with amine-functionalized silica, iPP/PP-g-MA/am-S200(5 %) the observed agglomerates become even smaller and show a bit looser structure than in nanocomposites with unmodified silica. Big agglomerates are seldom in this composition – cf. Fig. 4c, f.
Nanocomposites with S50 nanosilica (<d > =40 nm) also demonstrate aggregation and agglomeration of particles, yet the agglomerates observed here appear smaller than in composites with S200: the typical size is approx. 100-500 nm in all S50 nanocomposites, although agglomerates in iPP/S50 (5 %) nanocomposite are found slightly larger than those in iPP/PP-g-MA/S50 (5 %) or iPP/PP-g-MA/am-S50 (5 %) nanocomposites – cf. high magnification micrographs in Fig. 5c, e and f. Additionally, the number of big agglomerates found in nanocomposites with S50 is notably smaller than observed in the compositions with S200 nanosilica.
The image analysis of TEM micrographs allowed to determine the size distributions of particle agglomerates and estimate their average sizes. The agglomerate size was expressed in terms of equivalent diameter ED = (4 A/π)1/2, a size feature derived from the object area, giving the diameter of a circle with the same area (A) as the measured object. The obtained distributions are presented as insets in the Figs. 4a–c and 5a–c. It can be noticed that for both S200 and S50 silica the agglomerate size distribution of iPP/SiO2 binary nanocomposites is the broadest, while the narrowest distribution is observed in the iPP/PP-g-MA/am-SiO2 hybrid systems. Along with narrowing the size distribution the average size of agglomerates changes from ED = 184 nm through 196 nm to 142 nm for iPP/S200 (5 %), iPP/PP-g-MA/S200 (5 %) and iPP/PP-g-MA/am-S200 (5 %), respectively. For nanocomposites with S50 silica the estimated values of the average agglomerate size are: 172 nm in iPP/S50 (5 %), 148 nm in iPP/PP-g-MA/S50 (5 %) and 156 nm in iPP/PP-g-MA/am-S50 (5 %).
The overall conclusion drawn from microscopic examination is that neither amine functionalization of silica surface nor use of PP-g-MA compatibilizer caused dramatic improvement of silica dispersion. The tendency to agglomeration was still observed, also in the iPP/PP-g-MA/am-SiO2 hybrid nanocomposite. The modification, however, resulted in reduction of the average size of agglomerates, and additionally in some loosening of their packing as well as almost complete elimination of very big agglomerates, presence of which can appear very detrimental for mechanical performance of composite. Therefore, the aminosilane surface modification of silica particles can be considered effective.
The morphology and structure of the matrix
Preliminary SEM observations (Fig. 3) suggested that all composites studied crystallized in the spherulitic fashion, similar to plain polypropylene. The spherulitic morphology was confirmed by TEM observations and additionally by SALS measurements - the Hv SALS patterns observed with crossed polarizers demonstrated a four-lobes shape, characteristic for spherulitic morphology (patterns not shown). Qualitative estimations of these patterns suggested that the average spherulite size decreases in nanocomposites with increasing silica loading.
In order to determine the structural details of the PP matrix the wide- and small angle X-ray scattering (WAXS, SAXS) and calorimetric (DSC) measurements were performed. The collected X-ray diffractograms (examples presented in Fig. 6), revealed basically the same structure of the crystalline phase in all samples studied. The main crystalline form present in both iPP and its nanocomposites is the monoclinic α modification. Additionally, a small fraction of crystallites of trigonal β modification was detected. The parameters of the structure, estimated on the basis of diffractograms are summarized in Table 3. The amount of β modification estimated for plain iPP was about 2.5 wt.%, null for iPP/SiO2 binary nanocomposites, 2.5–3 wt.% for iPP/PP-g-MA/SiO2 ternary nanocomposites, and increased up to 5–9 wt.% in hybrid nanocomposites with modified silica, iPP/PP-g-MA/am-SiO2. The increase of β -PP fraction in PP-silica nanocomposites is not much surprising - the ability of silica particles to nucleate preferentially the β -PP crystals upon nonisothermal crystallization was already reported in the literature [48]. The highest amount of β phase in nanocomposites with am-SiO2 particles is probably related to their better dispersion than unmodified SiO2 or specific structure with PP-g-MA chains concentrated at interfaces.

X-Ray difractorams of the PP/SiO2 nanocomposites (5 wt.% of S200 silica), samples obtained by compression molding followed by fast cooling
The overall crystallinity was estimated roughly from X-ray data with the peak fitting procedure - estimation with better accuracy would require diffraction data collected in significantly broader span 2Θ than in this study. Comparison with the DSC-based estimations, reported later, suggests that the values obtained from X-ray data are systematically overestimated. Nevertheless, the obtained results suggest a tendency of PP crystallinity to decrease in nanocomposites as compared to plain iPP (cf. Table 3). Also the average lateral size of crystallites along [110, 040] directions, determined using the Scherrer equation (columns 4 and 5), is in all nanocomposites noticeably smaller than the respective size in plain iPP crystallized at similar conditions. This indicates that silica particles, in spite of some activity as primary nucleation centers, disturb the growth of crystals of the PP matrix, resulting in lower perfection of the final crystalline structure.
Both plain iPP and nanocomposites, when probed with 2-D SAXS demonstrated scattering in the form of uniform ring, typical for non-oriented semicrystalline polymer. The long period (LP) was estimated from one-dimensional sections of collected 2-D SAXS patterns. It was calculated from the position of the maximum in background and Lorentz corrected curve using the Bragg’s law. The obtained values are reported in Table 3. It was found that all nanocomposites exhibit similar increase of the long period with addition of silica nanofiller, irrespective of its type and surface treatment. The observed changes in the long period reflect the changes in the lamellar structure of the PP matrix, mainly an increase of the lamella thickness (which manifests also in an increase of the temperature of the melting peak, reported later in this section) and reduction of crystallinity (suggested by WAXS), induced by presence of silica. Rough estimates of crystal thickness, based on SAXS/WAXS data, are given in the last column of Table 3.
The DSC results obtained in the heating-cooling-heating cycle are summarized in Table 4. Representative heating thermograms are depicted in Fig. 7. All thermograms demonstrate melting and crystallization peaks typical for the α modification. The heating thermograms of some nanocomposites, especially those containing above 2.5 % of S200 silica, exhibit additionally a small peak located at the shoulder of the α melting peak, around 145 °C, cf. Fig. 7. This peak corresponds to melting of a small fraction of β crystals, which observation is consistent with results of X-ray diffraction, discussed earlier.

DSC thermograms recorded during 2nd heating with the rate of 10 °C/min of iPP and its nanocomposites containing 2.5 wt.% of silica
The data presented in Table 4 show that the melting (in both first and second heating cycle) and crystallization temperatures of iPP/SiO2 and iPP/PP-g-MA/SiO2 nanocomposites with unmodified silica remain nearly constant or increase slightly (less than 1 °C) with increasing content of silica, either S50 or S200 type. Similarly, only a relatively small increase in crystallinity is observed with increasing silica content. These observation suggest that the presence of unmodified silica in a nanocomposite influences crystallization process of PP matrix in a rather limited extent. Comparing nanocomposites with S50 and S200 it can be inferred that S200 silica of higher specific surface influences PP crystallization somewhat stronger than S50 – temperature of the crystallization peak, Tc, slowly increases with concentration in nanocomposites containing S200 while remains practically constant in those with S50. In general, the influence of unmodified silica on PP crystallization can be assessed as relatively low. In contrast, the surface-modified nanosilica, both am-S200 and am-S50, shows notably higher ability to modify the crystallization of the PP matrix. It manifests in significant increase of the crystallization peak temperature, by approx. 7-8 °C at 5 wt.% of nanofiller, either am-S200 or am-S50. That increase probably results from enhanced primary nucleation, i.e. am-SiO2 acts as a nucleation agent for polypropylene. This conclusion is confirmed by SALS results, reported earlier, that suggest smaller average spherulite size in nanocomposites than in plain iPP. As a consequence of crystallization proceeding at higher temperature, lamellae in nanocomposites with am-SiO2 grow thicker than in plain iPP or nanocomposites with unmodified SiO2, and hence show an elevated melting temperature (by 1-2 °C, cf. Table 4). The other effect of activity of am-SiO2 suggested by DSC data is an increase of the degree of PP crystallinity in nanocomposites, observed clearly, especially in the second heating run after crystallization at unified conditions. This result is opposite to X-ray based crystallinity rough estimations, suggesting some reduction of PP crystallinity in nanocomposites containing 5 wt.% of silica (Table 2). However, as already mentioned, an accuracy of X-ray estimations was limited.
Thermal stability
Thermal and thermo-oxidative stability of PP/silica nanocomposites was studied by thermogravimetric analysis (TGA) in an inert (N2) and oxidative (air) atmosphere, respectively. TGA weight loss curves in nitrogen are presented in Fig. 8a, while those obtained in air in Fig. 8b. Temperatures of the maximum loss rate and the residue weights are reported in Table 5.

TGA weight loss curves of nanocomposites, all containing 5 wt.% of S200 silica, heated in the nitrogen (a) and air (b) atmosphere. Insets show the derived curves of the weight loss rate
When heated in inert nitrogen atmosphere weight loss curves of PP and nanocomposites are similar in shape, typical for thermal degradation of polypropylene. Both silica grades, plain or modified, used here increase the temperature of the maximum weight loss rate of PP, from 4 to 8 °C, cf. Table 5, demonstrating a moderate thermal stabilization of PP matrix. The strongest stabilization effect was observed in hybrid nanocomposites with am-S200 amine-modified silica. The residue left in nanocomposites heated up to 600 °C is about 5 wt.%, which coincides well with the silica loading.
Heating in an oxidative atmosphere (air) leads to significantly faster degradation of iPP than in inert conditions due to peroxidation [49]. It results in a substantial decrease of temperature of degradation of iPP by nearly 100 °C comparing to heating in N2 atmosphere. Samples of PP/silica nanocomposites demonstrate a highly improved thermo-oxidative stability comparing to plain iPP, which manifests in the shift of the maximum of weight loss rate from 363 °C (plain iPP) to 397-405 °C for nanocomposites containing 5 wt% of silica. The greatly improved thermo-oxidative stability of PP/silica nanocomposites can be explained by the accumulation of silica nanoparticles on the sample surface during the earlier stage of nanocomposite degradation, allowing the formation of a ceramic layer, which can act as a protective barrier, limiting the polymer volatilization rate [50]. This effect is slightly stronger in iPP/PP-g-MA/SiO2 and iPP/PP-g-MA/am-SiO2 than in the binary composite iPP/SiO2 due to the improved silica dispersion that results in formation of more uniform and tight protective layer of nanoparticles accumulated at the specimen surface.
Mechanical performance
Tensile properties
Figure 9 shows exemplary nominal stress – nominal strain curves of selected nanocomposites (each containing 1.25 wt.% of S200 silica particles) determined in tensile test at room temperature. The most relevant mechanical properties: the elastic modulus (E), yield stress (σy) and elongation at break (εb), determined from the curves are summarized in Fig. 10. All samples demonstrated typical cold-drawing behavior with clear neck formation and stress-whitening of the necked part of the specimen, related to cavitation, which is common in tensile deformation of semicrystalline polymers [51]. Most of the compositions studied fractured early after formation of the neck, at relatively low strain, not exceeding 20 %. Those samples which deformed to higher strains usually did not show distinct strain hardening prior to a break. Therefore, the stress at yield actually represents the tensile strength of the materials studied. As it is frequently reported for polyolefin-based nanocomposites [4, 5, 15–28], introduction of silica nanoparticles into the system leads to an increase of the elastic modulus. We also observed this reinforcing effect in nanocomposites studied here - the modulus of nanocomposites increased with increasing silica content, up to about 12 % in nanocomposites filled with 5 wt.% of silica, cf. Fig. 10a, b. An increase of elastic modulus in nano-filled samples is explained in literature as a consequence of stiffness of particles and presence of an interphase layer around nanoparticles improving the stress transfer across interface that, in turn, allows larger participation of stiff particles in an accommodation of tensile strain. It is also frequently postulated that the particles can additionally restrict the mobility of polymer chains within such interfacial layers of the matrix. As it will be discussed later this is also the case of the studied PP/silica nanocomposites.

Representative nominal stress-nominal strain curves obtained in tensile experiment of iPP and its nanocomposites with 1.25 wt.% of plain or modified S200 silica. Only an initial part of the curves, up to ε = 50 %, is shown

Tensile properties of iPP and iPP/PP-g-MA/SiO2 or iPP/PP-g-MA/am-SiO2 nanocomposites with S200 (a, c, e) or S50 (b, d, f) silica: (a, b) Young modulus; (c, d) stress at yield; (e, f) ultimate strain
The reinforcing effect is the weakest in the binary composites iPP/SiO2, moderate in iPP/PP-g-MA/SiO2 and the strongest in iPP/PP-g-MA/am-SiO2 hybrid nanocomposites. Generally, the modulus increases more in nanocomposites with S200 than those with S50, especially at low nanofiller content. This can be related to higher specific surface area of S200 than S50 implying larger volume of the PP interfacial layers, likely to be stiffened, in the nanocomposites containing S200 than in those with S50. The relatively stronger increase of stiffness is observed at low concentration of the nanofiller, probably due to better silica dispersion (fewer big agglomerates). As already discussed, using the compatibilizer as well as surface-modified particles results in an improved dispersion, which in turn leads to an increase of the modulus of modified systems compared to the simple iPP/SiO2 binary composite. Additionally, these modifications enhance stress transfer between PP matrix and particles owing chemical bonding of PP chains to a particle, which also contributes to an increase of the nanocomposite modulus. Moreover, the primary loose filler aggregates, characteristic for fumed silica and present in all nanocomposites studied, provide another reinforcement mechanism, postulated by Dorigato et al. [52].
Along with increasing stiffness a slight increase of tensile strength, equivalent to the yields stress σy, with increasing silica content was observed (Fig. 10c, d). From a general point of view, an increase of the tensile yield stress of composites, even small, is considered as an indication of a relatively strong filler-matrix interaction, otherwise σy would rather decrease with the introduction of the filler [53–55]. In fact, Galeski et al. [56] reported that in polyolefins filled with traditional micro-filler an increase in the elastic modulus of the material is accompanied by a heavy decrease of the yield stress, because traditional fillers weakly adhered to the matrix hardly bear the load in the direction of tension. On the other hand, an increase of the yield stress in the studied nanocomposites can be also a result of increasing lamella thickness, l c (cf. Table 3) as the yield stress is known to depend on the crystal thickness, σy∝exp (−1/l c ) [57]. The yielding process and formation of the neck takes longer in nanocomposites than in plain iPP, demonstrating a narrow yield tooth in the stress-strain curve. This effect is probably associated with slightly diffused and broader zone of plastic deformation in PP/silica nanocomposites as compared to plain iPP.
In spite of weaker localization, the deformation of nanocomposites is generally unstable and samples break usually at low strains, below 20–30 %. This is the case of practically all iPP/SiO2 and iPP/PP-g-MA/SiO2 nanocomposites with unmodified silica (of both grades). In contrast, ternary hybrid nanocomposites containing surface modified silica iPP/PP-g-MA/am-SiO2 show much higher ductility, observed especially at low content of silica, 1.25 wt.%. Comparing nanocomposites containing am-S200 and am-S50 silica one can find a better performance of the latter. All above observations can be explained on the morphological ground: TEM study demonstrated that nanocomposites with untreated silica contained a significant number of big agglomerates, which upon tension act as critical sized flaws that trigger fracture phenomena, leading quickly to premature failure. When number of such detrimental agglomerates becomes low, as in iPP/PP-g-MA/am-SiO2 composites at low silica concentration, the iPP matrix is allowed to deform to much higher strain, comparable or higher than attained by plain iPP.
The above reported semi-brittle behavior is typical for numerous nanocomposites. In fact, in most of works on polyolefin nanocomposites, the stiffening effect due to nanoparticles is accompanied by a heavy embrittlement, with frequently dramatic reduction of the elongation to break [5, 54]. The toughening effect induced by aminosilanized silica nanoparticles at low concentrations can be considered an interesting result, in line with results reported for PE-fumed silica nanocomposites [58, 59]. It is likely that the good dispersion of silica aggregates leads to relatively low stress concentrations and hence reduce crack nucleation phenomena.
Dynamic properties
In order to find out any influence of silica particles on mobility of neighboring PP chains the dynamic mechanical analysis (DMA) was performed. Figure 11 presents the temperature dependencies of the storage and loss moduli, E’ and E”, respectively, determined for plain iPP and nanocomposites. It can be observed that the presence of silica particles in nanocomposites leads to the stiffening of the polypropylene matrix, manifesting in higher storage modulus E’ of nanocomposites than plain iPP in the entire temperature range explored. The largest increase in E’ is observed in the binary composites iPP/SiO2, while nanocomposites compatibilized with PP-g-MA demonstrate only a moderate increase of E’. When analyzing the loss modulus curves E” one can distinguish three maxima related to the relaxation transitions typical for polypropylene: a very low intensity γ at approx. –40 °C (sub-segmental and side-chain relaxations), β at approx. 8-10 °C (corresponding to the glass transition within the amorphous phase) and α around 60-66 °C (the effect of complex changes occurring both in crystals and adjacent disordered regions) [60, 61]. Since the observed peaks of α and β relaxation are broad and overlap each other their positions were determined with the peak fitting procedure. The estimated values are given in Table 6. It was found that both relaxation processes in nanocomposites are affected by presence of silica particles, especially in iPP/PP-g-MA/am-SiO2 hybrid nanocomposites, in which PP chains were grafted onto silica surface through MA and NH2 functional groups: the maximum of β shifts up, from 8.1 °C in plain iPP to 10.3 °C in iPP/PP-g-MA/am-SiO2 hybrids, while the maximum of α relaxation peak moves down, from 67.3 °C to 60–62 °C, respectively. The iPP/SiO2 and iPP/PP-g-MA/SiO2 nanocomposites with unmodified silica show similar, although significantly smaller changes in relaxation temperatures. The observed variation of relaxation temperatures is systematic, observed in all nanocomposites. There is practically no difference between respective nanocomposites containing S200 and S50 silica particles. The increase of the glass transition temperature (β peak) demonstrates that presence of silica particles in nanocomposites leads to a certain reduction of mobility of PP chains neighboring particles, especially in iPP/PP-g-MA/am-SiO2 hybrid nanocomposites with PP chains grafted onto surface of particles. This result confirms that restricted chain mobility can be one of the sources of enhanced elastic modulus and yield strength in nanocomposites.

The dependencies of the storage (E’) and loss (E”) moduli on temperature determined by DMA in the single cantilever bending mode for PP/silica nanocomposites containing 5 wt.% of S200 silica particles
Impact properties
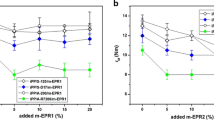
Impact properties of nanocomposites were probed at room temperature in two independent tests: Izod impact test (a notched beam deformed in bending) and tensile impact test (extension of a dumb-bell unnotched sample). All samples, including plain iPP demonstrated a quasi-brittle behavior in both tests. Any macroscopic plastic zone was nearly not observed. As shown in Fig. 12a, b the Izod impact strength of the iPP/SiO2 and iPP/PP-g-MA/SiO2 nanocomposites remain nearly constant irrespective of silica concentration, yet slightly below the impact strength of plain iPP. Moreover, nanocomposites with S50 silica show the Izod impact resistance slightly lower than those with S200. The differences are small, practically within the range of experimental error. On the contrary, the iPP/PP-g-MA/am-SiO2 hybrid nanocomposites demonstrate the Izod impact resistance undoubtedly higher than iPP, and increasing with increasing concentration of nanosilica of both am-S50 and am-S200 grade. The Izod impact strength of iPP/PP-g-MA/am-SiO2 is nearly 70 % larger than that of plain iPP. That increase of impact energy is accompanied by an increase of the maximum force recorded at impact. Samples of iPP/PP-g-MA/am-SiO2 demonstrate the highest values of the maximum force, cf., Fig. 12c, d.

The impact strength (a, b) and the maximum force (c, d) determined in the Izod impact test of notched samples of PP nanocomposites with S200 (a, c) and S50 (b, d) silica
Similar impact characteristics were observed also in the tensile impact – see Fig. 13. Since the samples were not notched and moreover, loaded in a different mode than in Izod test, the impact strength determined is much different, actually higher than that found in the Izod test. The same comment holds for the force measured in tensile impact and Izod tests. Nevertheless, practically the same trends as in results of the Izod test can be recognized here: the iPP/PP-g-MA/am-SiO2 hybrid nanocomposites with both am-S50 and am-S200 nanosilica exhibit the highest toughness.

The impact strength (a, b) and the maximum force (c, d) in tensile impact test of unnotched samples of PP nanocomposites with S200 (a, c) and S50 (b, d) silica
The impact strength (either Izod or tensile) of iPP/PP-g-MA/am-SiO2 nanocomposite is significantly improved probably due to reinforcing effect of relatively well dispersed silica (higher stress). Unfortunately, the effect was not strong enough to activate widespread plastic deformation, which would be a necessary condition for a super-tough material response [62]. The improved toughness was found in the entire range of composition in spite of low ultimate strain in tensile tests at low speed, observed at 2.5–5 wt.% filler loading. This can be explained by the difference in size of deformation region – the entire gauge length in tensile samples and the narrow deformation zone in front of the notch in Izod. Drawability in tension is presumably reduced by presence of big agglomerates within the deformation region. Even a single critical-sized agglomerate can trigger a premature fracture. At higher filler loadings probability of finding of such a critical agglomerate within large deforming part of a tensile specimen is much higher than in the very narrow deformation zone of the Izod sample. This in turn, causes low ductility in tension in spite of enhanced impact toughness.
Conclusions
The following conclusions can be drawn from the study reported:
-
According to NMR results a significant fraction of amine group in amine-modified silica particles is located on the top of the modification layer around particles, thus are easily accessible for further reactions upon compounding with a polymer.
-
Microscopic examination revealed that neither amine functionalization of silica surface nor use of PP-g-MA compatibilizer caused dramatic improvement of silica dispersion. The tendency to agglomeration was still observed, also in the iPP/PP-g-MA/am-SiO2 hybrid nanocomposites. The applied modification, however, resulted in reduction of the average size of agglomerates and additionally in some loosening of their packing as well as almost complete elimination of big agglomerates, presence of which can appear very detrimental for mechanical performance of a composite. Therefore, the aminosilane surface modification of silica particles can be considered as effective.
-
Polypropylene crystallizes in a spherulitic fashion in both plain iPP and nanocomposites. Silica particles demonstrate some nucleation activity towards iPP that manifests in an increase of the crystallization temperature, up to approx. 7-8 °C at 5 wt.% of nanofiller, and reduction of the average spherulite size. The surface-modified nanosilica particles demonstrate higher ability to modify the crystallization of the PP matrix than bare silica. As a result of crystallization proceeding at higher temperature, lamellae in nanocomposites with am-SiO2 grow thicker than in plain iPP or nanocomposites with unmodified SiO2, and hence show an elevated melting temperature. The other effect of activity of am-SiO2 is an increase of the degree of PP crystallinity in nanocomposites, observed clearly in the second heating run in DSC. On the other hand, presence of silica particles disturb the growth of crystallites, which results in less perfect structure consisting of smaller crystallites than observed in plain iPP.
-
Samples of iPP/silica nanocomposites demonstrate highly improved thermo-oxidative stability that manifests in the upward shift of the maximum of weight loss rate by approx. 40 °C, observed in nanocomposites with 5 wt% of silica. The greatly improved thermo-oxidative stability of iPP/silica nanocomposites can be explained by the accumulation of silica nanoparticles on the sample surface during the earlier stage of nanocomposite degradation, allowing the formation of a ceramic protective layer, limiting the polymer volatilization rate. The effect is the strongest in the iPP/PP-g-MA/am-SiO2 hybrid nanocomposite due to improved particle dispersion.
-
Nanocomposites with silica demonstrate enhanced stiffness and the yield strength comparing to plain iPP. That increase is a result of the interphase layer around nanoparticle, improving the stress transfer across interface and allowing larger participation of stiff particles in an accommodation of tensile strain. Additionally, the mobility of PP chains within interfacial layers of the matrix is reduced. Use of surface-modified particles and compatibilizer results in an improved dispersion as well as stronger interactions within interfacial layer, which in turn leads to an increase of the stiffness and strength of modified systems as compared to the simple iPP/SiO2 composite.
-
In most of nanocomposites studied, except iPP/PP-g-MA/am-SiO2 hybrid nanocomposite at low silica loading, the ductility is nearly lost. This is related to big silica agglomerates which act as critical-sized flaws and bring a premature material failure. However, the dispersion of nanofiller in iPP/PP-g-MA/am-SiO2 nanocomposites is much improved and there is much less agglomerates than in other composites tested, especially in compositions with low silica content. Owing to an absence of large agglomerates deformability of such nanocomposites is not lost, and remains comparable or even become better than plain iPP. It is likely that the good dispersion of silica aggregates leads to relatively low stress concentrations and hence reduce crack nucleation phenomena.
-
The iPP/PP-g-MA/am-SiO2 hybrid nanocomposite shows an improved impact toughness as compared to plain iPP (by nearly 70 % in the Izod test) or other nanocomposites. Changes in energy absorbed at impact are accompanied by an increase of the maximum force recorded at impact with increasing content of silica and surface modification. Most probably, enhancement of the impact strength is due to reinforcing effect of relatively well dispersed silica particles. Unfortunately, the effect is not strong enough to activate widespread plastic deformation in a sample, which would be a necessary condition for a super-tough material response.
References
Paul DR, Robeson LM (2008) Polymer nanotechnology: nanocomposites. Polymer 49(15):3187–3204
Njuguna J, Pielichowski K, Desai S (2008) Nanofiller-reinforced polymer nanocomposites. Polym Adv Technol 19(8):947–959
Pavlidou S, Papaspyrides CD (2008) A review on polymer–layered silicate nanocomposites. Prog Polym Sci 33(12):1119–1198
Zhang MQ, Rong MZ, Ruan WH (2009) Nanoparticles/polymer composites: fabrication and mechanical properties. In: Karger-Kocsis J, Fakirov S (eds) Nano- and micro-mechanics of polymer blends and composites. Hanser, Munich, pp. 93–140
Zou H, Wu S, Shen J (2008) Polymer/silica nanocomposites: preparation, characterization, properties, and applications. Chem Rev 108(9):3893–3957
Sun LY, Gibson RF, Gordaninejad F, Suhr J (2009) Energy absorption capability of nanocomposites: a review. Compos Sci Technol 69(14):2392–2409
Choudalakis G, Gotsis AD (2009) Permeability of polymer/clay nanocomposites: a review. Eur Polym J 45(4):967–984
Zhao C, Qin H, Gong F, Feng M, Zhang S, Yang M (2005) Mechanical, thermal and flammability properties of polyethylene/clay nanocomposites. Polym Degrad Stab 87(1):183–189
Michler GH, Balta-Calleja FJ (2012) Nano- and micromechanics of polymers. Carl Hanser Verlag, Munich
Iler RK (1979) The chemistry of silica. Wiley, New York
Kim HC, Dubois G (2005) Dekker encyclopedia of nanoscience and nanotechnology. Taylor & Francis, New York
Jana SC, Jain S (2001) Dispersion of nanofillers in high performance polymers using reactive solvents as processing aids. Polymer 42:6897–6905
Nalwa HS (ed) (2003) Handbook of organic-inorganic hybrid materials and nanocomposites. American Scientific Publishers, Stevenson Ranch
Pegoretti A (2009) Creep and fatigue behaviour of polymer nanocomposites. In: Karger-Kocsis J, Fakirov S (eds) Nano- and micro-mechanics of polymer blends and composites. Hanser, Munich, pp. 301–339
Wu CL, Zhang MQ, Rong MZ, Friedrich K (2002) Tensile performance improvement of low nanoparticles filled-polypropylene composites. Compos Sci Technol 62(10–11):1327–1340
Bikiaris DN, Vassiliou A, Pavlidou E, Karayannidis GP (2005) Compatibilisation effect of PP-g-MA copolymer on iPP/SiO2 nanocomposites prepared by melt mixing. Eur Polym J 41(9):1965–1978
Bikiaris DN, Papageorgiou GZ, Pavlidou E, Vouroutzis N, Palatzoglou P, Karayannidis GP (2006) Preparation bv melt mixing and characterization of isotactic polypropylene/SiO2 nanocomposites containing untreated and surface-treated nanoparticles. J Appl Polym Sci 100(4):2684–2696
Bouaziz A, Jaziri M, Dalmas F, Massardier V (2014) Nanocomposites of silica reinforced polypropylene: correlation between morphology and properties. Polym Eng Sci 54(9):2187–2196
Chen JH, Rong MZ, Ruan WH, Zhang MQ (2009) Interfacial enhancement of nano-SiO2/polypropylene composites. Compos Sci Technol 69(2):252–259
Lin OH, Mohd Ishak ZA, Akil HM (2009) Preparation and properties of nanosilica-filled polypropylene composites with PP-methyl POSS as compatibiliser. Mater Des 30(3):748–751
Rong MZ, Zhang MQ, Pan SL, Friedrich K (2004) Interfacial effects in polypropylene-silica nanocomposites. J Appl Polym Sci 92(3):1771–1781
Studziński M, Jeziórska R, Szadkowska A, Zielecka M (2014) Modified nanosilica-filled polypropylene composites with glycidyl methacrylate grafted ethylene/n-octene copolymer as compatibilizer. Polimery 59:625–635
Garcia M, van Vliet G, Jain S, Schrauwen BAG, Sarkissov A, van Zyl WE, Boukamp B (2004) Polypropylene/SiO2 nanocomposites with improved mechanical properties. Rev Adv Mater Sci 6(2):169–175
Papageorgiou DG, Vourlias G, Bikiaris DN, Chrissafis K (2014) Effect of silica nanoparticles modification on the thermal, structural, and decomposition properties of a β-nucleated poly(propylene-co-ethylene) matrix. Macromol Chem Phys 215(9):839–850
Pavlidou E, Bikiaris D, Vassiliou A, Chiotelli M, Karayannidis G (2005) Mechanical properties and morphological examination of isotactic polypropylene/SiO2 nanocomposites containing PP-g-MA as compatibilizer. J Phys Conf Ser 10(1):190–193
Zhou RJ, Burkhart T (2011) Polypropylene/SiO2 nanocomposites filled with different nanosilicas: thermal and mechanical properties, morphology and interphase characterization. J Mater Sci 46(5):1228–1238
Zoukrami F, Haddaoui N, Vanzeveren C, Sclavons M, Devaux J (2008) Effect of compatibilizer on the dispersion of untreated silica in a polypropylene matrix. Polym Int 57(5):756–763
Taniike T, Toyonaga M, Terano M (2014) Polypropylene - grafted nanoparticles as a promising strategy for boosting physical properties of polypropylene-based nanocomposites. Polymer 55(4):1012–1019
Karian HG (ed) (2003) Handbook of polypropylene and polypropylene composites. Plastics Engineering. Marcel Dekker, New York
Rong MZ, Zhang MQ, Zheng YX, Zeng HM, Walter R, Friedrich K (2001) Structure–property relationships of irradiation grafted nano-inorganic particle filled polypropylene composites. Polymer 42(1):167–183
Boyer C, Boutevin B, Robin JJ (2005) Study of the synthesis of graft copolymers by a reactive process. Influence of the copolymer structure on the adhesion of polypropylene onto poly(vinylidene fluoride). Polym Degrad Stab 90(2):326–339
Fina A, Tabuani D, Peijs T, Camino G (2009) POSS grafting on PPgMA by one-step reactive blending. Polymer 50(1):218–226
Fina A, Monticelli O, Camino G (2010) POSS-based hybrids by melt/reactive blending. J Mater Chem 20(42):9297–9305
Grala M, Bartczak Z, Pracella M (2013) Morphology and mechanical properties of polypropylene-POSS hybrid nanocomposites obtained by reactive blending. Polym Compos 34(6):929–941
Krigbaum WR, Uematsu I (1965) Heat and entropy of fusion of isotactic polypropylene. J Polym Sci A Polym Chem 3(2):767–776
Wojdyr M (2010) Fityk: a general-purpose peak fitting program. J Appl Crystallogr 43:1126–1128
Turner-Jones A, Aizlewood JM, Beckett DR (1964) Macromol Chem 75:134–158
Alexander LE (1969) X-ray diffraction methods in polymer science. Wiley-Interscience, New York
Kurth DG, Bein T (1992) Quantification of the reactivity of 3-aminopropyl-triethoxysilane monolayers with the quartz-crystal microbalance. Angew Chem Int Ed Engl 31(3):336–338
Kurth DG, Bein T (1993) Surface reactions on thin layers of silane coupling agents. Langmuir 9(11):2965–2973
Ek S, Iiskola EI, Niinistö L (2004) Atomic layer deposition of amino-functionalized silica surfaces using N-(2-aminoethyl)-3-aminopropyltrimethoxysilane as a silylating agent. J Phys Chem B 108(28):9650–9655
Sen T, Bruce IJ (2012) Surface engineering of nanoparticles in suspension for particle based bio-sensing. Sci Report 2:564
van de Waterbeemd M, Sen T, Biagini S, Bruce IJ (2010) Surface functionalisation of magnetic nanoparticles: quantification of surface to bulk amine density. Micro Nano Lett 5(5):282
Bruce IJ, Sen T (2005) Surface modification of magnetic nanoparticles with alkoxysilanes and their application in magnetic bioseparations. Langmuir 21(15):7029–7035
Choi S-H, Cai Y, Newby B-MZ (2007) Stability Enhancement Of Polystyrene Thin Films On Aminopropyltriethoxysilane Ultrathin Layer Modified Surfaces. In: Silanes and Other Coupling Agents, Volume 4.
Wåhlander M, Nilsson F, Larsson E, Tsai W-C, Hillborg H, Carlmark A, Gedde UW, Malmström E (2014) Polymer-grafted Al2O3-nanoparticles for controlled dispersion in poly(ethylene-co-butyl acrylate) nanocomposites. Polymer 55(9):2125–2138
van Blaaderen A, Vrij A (1993) Synthesis and characterization of monodisperse colloidal organo-silica spheres. J Colloid Interface Sci 156(1):1–18
Pedrazzoli D, Pegoretti A, Kalaitzidou K (2015) Understanding the effect of silica nanoparticles and exfoliated graphite nanoplatelets on the crystallization behavior of isotactic polypropylene. Polym Eng Sci 55(3):672–680
Grassie N, Scott G (1985) Polymer degradation and stabilization. Cambridge University Press, Cambridge
Fina A, Tabuani D, Carniato F, Frache A, Boccaleri E, Camino G (2006) Polyhedral oligomeric silsesquioxanes (POSS) thermal degradation. Thermochim Acta 440(1):36–42
Pawlak A, Galeski A, Rozanski A (2014) Cavitation during deformation of semicrystalline polymers. Prog Polym Sci 39(5):921–958
Dorigato A, Dzenis Y, Pegoretti A (2013) Filler aggregation as a reinforcement mechanism in polymer nanocomposites. Mech Mater 61:79–90
Ahmed S, Jones FR (1990) A review of particluate reinforcement theories for polymer composites. J Mater Sci 25:4933–4942
Nielsen LE, Landel RF (1994) Mechanical properties of polymers and composites. M. Dekker, New York
Nicolais L, Nicodemo L (1973) Strength of particulate composite. Polym Eng Sci 13:469–477
Galeski A (2003) Strength and toughness of crystalline polymer systems. Prog Polym Sci 28(12):1643–1699
Brooks NWJ, Mukhtar M (2000) Temperature and stem length dependence of the yield stress of polyethylene. Polymer 41:1475–1480
Dorigato A, D’Amato M, Pegoretti A (2012) Thermo-mechanical properties of high density polyethylene – fumed silica nanocomposites: effect of filler surface area and treatment. J Polym Res 19(6)
Kontou E, Niaounakis M (2006) Thermo-mechanical properties of LLDPE/SiO2 nanocomposites. Polymer 47(4):1267–1280
Boyd RH (1985) Relaxation processes in crystalline polymers: experimental behaviour — a review. Polymer 26(3):323–347
Boyd RH (1985) Relaxation processes in crystalline polymers: molecular interpretation — a review. Polymer 26(8):1123–1133
Bartczak Z, Galeski A (2014) Mechanical properties of polymer blends. In: Utracki LA, Wilkie CA (eds) Polymer blends handbook, vol 2, 2nd edn. Springer Science+Business Media, Dordrecht, pp. 1203–1297
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Grala, M., Bartczak, Z. & Różański, A. Morphology, thermal and mechanical properties of polypropylene/SiO2 nanocomposites obtained by reactive blending. J Polym Res 23, 25 (2016). https://doi.org/10.1007/s10965-015-0914-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-015-0914-0