
Abstract
A symmetric photo-active trithiocarbonate was designed and synthesized, which can act not only as a functional reversible addition-fragmentation chain transfer (RAFT) agent, but also as a crosslinker in crosslinking poly(methyl methacrylate-co-hydroxy ethyl acrylate) (poly(MMA-co-HEA)) due to interactions of benzophenone moieties of trithiocarbonate and hydroxyl groups of poly(MMA-co-HEA) under UV irradiation. In addition, the crosslinked poly(MMA-co-HEA) demonstrates photo-promoted self-healing properties by reshuffling C-S bonds of trithiocarbonate.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Self-healing and self-repair phenomena are very common in living organisms. For example, a slight injury of our skins, fleshes or even bones can be self-healed within a period of time. The self-healing process of living organisms is sophisticated and elegant with several mechanisms involved [1]. Inspired by the natural self-healing process and with the purpose to improve lifespan, durability and reliability of materials especially those used in hardly accessible or dangerous places, self-healing of synthetic materials has drawn considerable attention in the past decades both in academia and in industry [2–4]. In fact, self-healing research progress has been reviewed from different perspectives of many papers in recent years [5–15].
There are mainly two kinds of methods for preparing self-healing materials according to principles, and one is use of self-healing agent usually as microcapsules embedded in material matrix [16–18], the other is based on reversible interactions of molecules to form dynamic bonds [19–24]. For the former approach, a crack of materials can break self-healing-agent-containing microcapsules and cause the self-healing-agent to flow into the crack by capillary effect and then solidify usually catalyzed by an embedded catalyst. This kind of repairing is relatively easy to realize, but the healing process can be usually performed only once, thus limiting its applications to a certain extent. By comparison, the latter method utilizes reversible or dynamic bonds to form thermoplastic or thermoset materials, and these bonds can be cleaved/reformed more than once with or even without external stimuli. The external stimuli are usually light, heat, electricity, mechanics, acids/bases, redox, etc. For example, photo-triggered reversible [2 + 2] cycloaddition of coumarins and [4 + 4] cycloaddition of anthracenes have been applied to produce self-healing materials [25, 26]. Chuo et al. has reported electrically induced self-healing polymers based on reversible guest–host complexation of β-cyclodextrin and ferrocene [18]. Typical heat-responsive reaction is [4 + 2] cycloaddition of a diene and a dienophile, known as Diels-Alder (DA) reaction, and the addition products can undergo reversible cleavage reaction under elevated temperatures, namely retro Diels-Alder (RDA) reaction. The DA reaction has been widely used to design self-healing materials, usually applying maleimide and furan derivatives as reactants [27, 28]. For instance, Tian and coworkers designed an epoxy resin by using N,N-diglycidyl-furfurylamine (DGFA) and N,N’-(4,4′-diphenylmethane) bismaleimide (DPMBMI) as reactants, which showed not only similar mechanical properties as commercial epoxy resins, but also thermal remendability [29]. Similarly, Broekhuis’s research group made use of Paal-Knorr reaction between polyketones and furfurylamine to prepare furan-functionalized polypyrrole, which further underwent DA and RDA reaction with dimaleimide to form the desired self-healing polymers [30]. Acids/bases-sensitive (pH sensitive) materials usually contain imine or acyl hydrazone groups, which can conduct reversible reactions under certain pH values [31]. A typical example is the work of Ono and coworkers, where bis-hydrazides and dialdehydes reacted to form poly(acyl hydrazone)s, and an acid catalyst can trigger acyl hydrazone’s bond exchange [32]. A similar work was reported by Deng et al., who condensed PEO-based bis-hydrazide with trialdehyde to afford a crosslinked polymer with reversible sol–gel transition and self-healing properties, and the critical apparent pH value of the gel is 4 [24]. Disulfide bond is typically redox-responsive, though it can also be cleaved by thermal scission, mechanical stress, and photo-irradiation [33–36]. A representative example is the work of Yoon and his colleagues, where atom transfer radical polymerization (ATRP) technique was used to synthesize a star-like macroinitiator, followed by synthesis of S-S bond-containing gel via chain extension, and this gel can be reversibly transformed into sol under reduction conditions. The prepared materials showed a rapid self-healing behavior without external triggers [36]. More recently, there are also dual responsive self-healing polymers that are more adaptive to external environment [23, 37].
In addition to dynamic chemical bonds, there are also a type of self-healing suprapolymers composed of non-covalent dynamic bonds such as hydrogen bond and donor-acceptor interaction [21, 38–42]. For example, Burnworth and coworkers synthesized ligand-terminated poly(ethylene-co-butylene) by Mitsunobu reaction, which can then coordinate with Zn2+ to form a metallosupramolecular polymer. The metal-ligand part of the supramolecular polymer can be excited on exposure to UV light and caused reversible disengagement, thereby making the polymer have self-healing property [43]. Fox et al. designed electron-rich pyrenyl end-capped oligomer and electron-deficient naphthalene-diimide (NDI) oligomer, and the two oligomers were reinforced with 7.5 wt.% of cellulose nanocrystals (CNCs) to form a self-mendable nanocomposite material by π-π interactions [44].
Reversible addition-fragmentation chain transfer (RAFT) polymerization as one of the most important living radical polymerization methods has been widely used to prepare polymers with topological architectures or unique properties [45–50]. Herein, we firstly designed a new functional molecule, i.e., a symmetric benzophenone-containing trithiocarbonate. Secondly, linear poly(MMA-co-HEA) was prepared by using this benzophenone-containing trithiocarbonate as the RAFT chain transfer agent. Thirdly, the linear poly(MMA-co-HEA) was crosslinked under UV irradiation, which showed self-healing property presumably due to reversibly reshuffling of the trithiocarbonate groups.
Experimental
Materials
4-Methyl benzophenone, urea hydrogen peroxide (UHP), aqueous HBr (>40 %), CS2, NaOH, tetrabutyl ammonium bromide (TBAB) were all AR grade and used without any treatment as purchased from Shanghai Aladdin Industrial Corporation. Monomer hydroxyl ethyl acrylate (HEA) and methyl methacrylate (MMA) were treated with NaOH prior to use in order to remove inhibitors.
Characterization
Gel permeation chromatography (GPC) was conducted on an HP 1100 HPLC, equipped with a Waters 2414 refractive index detector and three Styragel HR 2, HR 4, HR 5 of 300 × 7.5 mm columns (packed with 5 mm particles of different pore sizes). The column packing allowed the separation of polymers over a wide molecular weight range of 500–1,000,000. THF was used as the eluent at a flow rate of 1 mL.min−1 at 40 °C. PMMA standards were used as the reference. Structures of trithiocarbonate and the copolymer poly(MMA-co-HEA) were characterized by1H NMR spectroscopy on a Bruker AV 400 MHz spectrometer. CDCl3 was used as the solvent. Monomer conversions were determined by gravimetry. Crosslinking experiments were finished on a RW-UV AH400a UV curing meter. SEM images were afforded from SSX-550 Scan Electron Microscope.
Synthesis of 4-bromomethyl benzophenone
4-Methyl benzophenone (1.25 g, 6.4 mmol) and UHP (1.2 g, 12.8 mmol) were dissolved in dichloromethane (30 mL) and were heated to reflux. Under irradiation of a 100-W bulb, aqueous HBr (1.4 mL, 6.9 mmol) was added dropwise within about 20 min, followed by refluxing for additional 40 min. The reaction mixture was cooled, and water was added, separated, and the organic phase was dried on anhydrous Na2SO4, filtered, and finally concentrated in vacuo to afford a white solid, which was recrystallized in anhydrous ethanol to give pure 4-bromomethyl benzophenone (1.60 g, 91 %). M.p. 109–110 °C. IR: ν (cm−1) 3040, 1668, 1610, 1463, 1419, 852, 599.1H NMR (CDCl3): δ 7.80–7.77 (m, 4H), 7.60–7.58 (m, 1H), 7.51–7.47 (m, 4H), 4.53 (s, 2H).13C-NMR (CDCl3): δ 196.0, 142.1, 137.5, 137.4, 132.6, 130.6, 130.0, 129.0, 128.4, 32.3.
Synthesis of bis(4-benzoylbenzyl) carbonotrithioate (3)
Carbon disulfide (4 mL), dichloromethane (6 mL), TBAB (0.05 g) and 20 % aqueous NaOH (10 mL) were mixed and stirred at room temperature for 10 min. Then 4-bromomethyl benzophenone (0.275 g, 1 mmol) was added and the mixture was stirred at room temperature overnight. The yellow mixture was separated, and the organic layer was washed three times with brine, dried with anhydrous Na2SO4, and concentrated in vacuo to give crude product which was further purified on silica gel column to obtain a light yellow oil (0.35 g, 71 %). IR: ν (cm−1) 3060, 3028, 1725, 1637, 1602, 1492, 1454, 1382, 1064, 767, 701.1H NMR (CDCl3): δ 7.79–7.75 (m, 8H), 7.59–7.57 (m 2H), 7.50–7.45 (m, 8H), 4.70 (s, 4H).13C-NMR (CDCl3): δ 221.5, 196.0, 139.9, 137.5, 137.0, 132.5, 130.5, 130.0, 129.2, 128.92, 128.86, 128.6, 128.3, 41.0.
Preparation of poly(MMA-co-HEA) via RAFT polymerization using bis(4-benzoylbenzyl) carbonotrithioate (3) as the chain transfer agent
To a 50 mL of three-necked flask were added bis(4-benzoylbenzyl) carbonotrithioate (3) (0.105 g, 0.2 mmol), AIBN (3.3 mg, 0.0201 mmol), MMA (2.0 g, 20 mmol), HEA (1.16 g, 10 mmol) and toluene (5 mL). The mixture was stirred at 65 °C for 4 h under N2 atm. Access methanol was added to precipitate the copolymer product, filtered to give solid product (2.01 g). The polymer product was characterized by GPC to afford Mn,GPC = 6086 ≈ 6.1 kDa, PDI = 1.48.
UV-curing of the poly(MMA-co-HEA) copolymer
Poly(MMA-co-HEA) (1.0 g, 0.164 mmol) and bis(4-benzoylbenzyl) carbonotrithioate (0.2 g, 0.402 mmol) were dissolved in THF (3 mL). The solution was spin-coated on glass plates, and then irradiated under UV light for a period of time.
Self-healing of the UV-cured poly(MMA-co-HEA)
The cured poly(MMA-co-HEA) coating was slightly cut by a razor to form a scratch with the width of c.a. 0.2–0.8 μm. Then the scratch part of the polymer film was irradiated by UV light with wave-number of 365 nm. After a certain period of time (e.g., 10 min), changes of the scratch of the polymer was observed with SEM images.
Results and discussion
Synthesis of bis(4-benzoylbenzyl) carbonotrithioate (3)
Trithiocarbonate RAFT chain transfer agents are usually synthesized from alkyl thio-alcohols, carbon disulfide and appropriate alkyl halides under basic conditions [48]. Symmetric trithiocarbonates can also be prepared by the above usual methods, but a preferable approach is a straight reaction reported by Aoyagi and Endo between a benzyl halide and carbon disulfide in the presence of a base, where most dibenzyl trithiocarbonates can be prepared in quantitative yields [51]. By modification of Aoyagi’s method, bis(4-benzoylbenzyl) carbonotrithioate (3), a novel photo-active trithiocarbonate, was synthesized in 71 % yield by using 4-bromomethyl benzophenone (2) as the substrate (Scheme 1).

Synthesis of bis(4-benzoylbenzyl) carbonotrithioate (3)
Preparation and crosslinking of poly(MMA-co-HEA) with bis(4-benzoylbenzyl) carbonotrithioate (3)
Benzophenone is a widely used type-II photoinitiator, which can be photo-chemically excited to become triplet state under UV irradiation. However, the excited state cannot straightly initiate radical polymerization, and it requires abstracting a labile hydrogen atom from a suitable hydrogen donor species (i.e., co-photoinitiator) such as amines, alcohols, thiols, etc. When an alcohol is used as a hydrogen donor, the formed radicals can combine to result in cross-linking [52].
Thus, the new compound bis(4-benzoylbenzyl) carbonotrithioate (3) that contains two benzophenone moieties is expected to be a photo-crosslinking agent for hydroxyl groups-containing polymers. To verify the idea, hydroxyl groups-containing polymer poly(MMA-co-HEA) was prepared via RAFT polymerization by using bis(4-benzoylbenzyl) carbonotrithioate (3) as the chain transfer agent (Scheme 2). The other purpose of the polymerization is to confirm that compound 3 can act as a RAFT chain transfer agent.

Synthesis of poly(MMA-co-HEA) via RAFT polymerization by using bis(4-benzoylbenzyl) carbonotrithioate (3) as the chain transfer agent
Characterization of poly(MMA-co-HEA) by1H NMR was shown in Fig. 1a and b demonstrated1H NMR spectrum of bis(4-benzoylbenzyl) carbonotrithioate (3) as contrast. Weak peaks at the range of 7.46–7.77 ppm represented aryl protons of benzophenone moiety of the polymer, and the integral values of the peaks were designated as 18 since there were two benzophenone groups at the end of the polymer, and integral values of other peaks were based on this value. The peaks at 3.6–4.1 ppm corresponded to O-CH 2CH 2-OH groups of HEA, whose integral value was about 118, meaning that there were 118/4 ≈ 30 of HEA units in the copolymer. The single peaks at around 3.6 ppm was the methoxy groups of MMA unit in the polymer, and 93 of protons at the single peaks indicated that there were 93/3 ≈ 31 of MMA units in the polymer. The ratio of copolymer composition between MMA and HEA units in the linear poly(MMA-co-HEA) was 31/30 (i.e., 1.03/1), which was less than the ratio of initial comonomer composition at 2/1, indicating that the reactivity of HEA was higher than that of MMA during solution RAFT polymerization. Thus, the number average molecular weight of poly(MMA-co-HEA) can be calculated as follows:

a 1H NMR spectrum of poly(MMA-co-HEA); b 1H NMR spectrum of bis(4-benzoylbenzyl) carbonotrithioate (3) as contrast
where M 3 was the molecular weight of compound 3, 116 was the molecular weight of HEA unit, 100 was that of MMA unit. Further characterization of poly(MMA-co-HEA) by GPC gives Mn,GPC as 6.1 kDa with polydispersity of 1.48. The relatively broad molecular distribution may be due to possible branching side reactions caused by difunctional HEA monomer.
With bis(4-benzoylbenzyl) carbonotrithioate (3) and poly(MMA-co-HEA) in hand, next, we began to study photo-crosslinking of the two species. In fact, poly(MMA-co-HEA) as a linear polymer is a viscous liquid, and it cannot be solidified or cured to form a film even under UV irradiation (Fig. 2a). When bis(4-benzoylbenzyl) carbonotrithioate (3) and poly(MMA-co-HEA) are mixed in THF and spin-coated on a glass substrate, a transparent film can be easily formed under UV irradiation for 10 min as shown in Fig. 2b. Under UV light, the benzophenone part of bis(4-benzoylbenzyl) carbonotrithioate (3) as a photoinitiator is excited and trapps a hydrogen atom from hydroxyl groups of poly(MMA-co-HEA), and the subsequent radical-radical combination leads to crosslinking of poly(MMA-co-HEA) (Scheme 3). In addition to this main crosslinking reaction, other radical-radical combinations such as between the RAFT agents and those between the poly(MMA-co-HEA) may also occur during UV irradiation. Hardness of the film is measured by special pencil, with results shown in Table 1. In the first 30 min, hardness of the film changes rapidly under UV light (i.e., from viscous liquid to a solid film with the hardness of 3H), which indicates that crosslinking reactions occurred mainly within the first half an hour. Further irradiation for another 30 min makes the hardness increase slowly from 3H to 4H. Note that the colorless polymer becomes gradually light brown during exposure to UV light, which may be ascribed to disadvantages of benzophenone photoinitiator moiety.

a Linear poly(MMA-co-HEA) as a viscous liquid. b Mixtures of bis(4-benzoylbenzyl) carbonotrithioate (3) and linear poly(MMA-co-HEA) can form a yellow solid film on a glass substrate after UV irradiation for 10 min

Crosslinking of poly(MMA-co-HEA) and bis(4-benzoylbenzyl) carbonotrithioate (3)
Self-healing of crosslinked poly(MMA-co-HEA) under UV irradiation
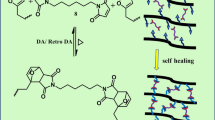
Trithiocarbonate acts as a crosslinker in the crosslinked poly(MMA-co-HEA) resin, furthermore, this special crosslinker may function as a chain transfer agent. Under UV irradiation, weak C-S single bonds of the trithiocarbonate may cleave to form the corresponding free radicals, which may re-link with each other in a way different from their previous connection (Scheme 4) [53].

Schematic illustration of cleavage and re-linking of the trithiocarbonate linkers with polymers. Note: “P” represents polymer, “S” is sulfur element
The self-healing process was demontrated by SEM photos in Fig. 3. Figure 3a is an undestroyed crosslinked poly(MMA-co-HEA) film, which is then cut by a razor to form a crack with width of 0.2–0.8 μm (Fig. 3b). The cut film is then irridiated for 5 min, and the cracks are mostly repaired (Fig. 3c). Further irridiation for another 5 min, the cracks are healed, with scars still existing (Fig. 3d).

Self-healing process of the crosslinked polymer films by SEM photographs: a a normal polymer film before cutting; b the polymer film after cutting by a razor, before UV irradiation; c the cut polymer film irradiated by UV for 5 min; d the cut polymer film irradiated by UV for 10 min
Conclusions
Benzophenone-containing symmetric trithiocarbonate was synthesized in moderate yield. Functionalities of this molecule lie in two aspects: one is the photo-active benzophenone moieties, the other is the trithiocarbonate part that is known as one of reversible addition-fragmentation chain transfer (RAFT) agents. The benzophenone moieties of the trithiocarbonate can act as a crosslinker of viscous linear poly(MMA-co-HEA) to form a thermoset copolymer resin film with hardness of up to 4H under UV irradiation for 60 min. Furthermore, the crosslinked poly(MMA-co-HEA) shows self-healing properties promoted by UV light, which may stem from reshuffling of weak and reactive C-S single bonds of trithiocarbonate under UV irradiation. SEM images indicate that a crack with width of 0.2–0.8 μm can be self-repaired within 10 min.
References
Amendola V, Meneghetti M (2009) Self-healing at the nanoscale. Nanoscale 1:74–88
García-Huete N, Laza JM, Cuevas JM, Gonzalo B, Vilas JL, León LM (2014) Shape memory effect for recovering surface damages on polymer substrates. J Polym Res 21:481
Wei Q, Wang J, Shen XY, Zhang XA, Sun JZ, Qin AJ, Tang BZ (2013) Self-healing hyperbranched poly(aroyltriazole)s. Sci Rep. doi:10.1038/srep01093
Rahimi A, Amiri S (2015) Self-healing hybrid nanocomposite coatings with encapsulated organic corrosion inhibitors. J Polym Res 22:624
Wojtecki RJ, Meador MA, Rowan SJ (2011) Using the dynamic bond to access macroscopically responsive structurally dynamic polymers. Nat Mater 10:14–27
Zhang MQ, Rong MZ (2012) Theoretical consideration and modeling of self‐healing polymers. J Polym Sci Part B Polym Phys 50:229–241
Wei Z, Yang JH, Zhou JX, Xu F, Zrínyi M, Dussault PH, Osada Y, Chen YM (2014) Self-healing gels based on constitutional dynamic chemistry and their potential applications. Chem Soc Rev. doi:10.1039/c4cs00219a
Zhang MQ, Rong MZ (2012) Design and synthesis of self-healing polymers. Sci Chin Chem 55:648–676
Li SL, Han P, Xu HP (2012) Self-healing polymeric materials. Prog Chem 24:1346–1352 (in Chinese)
Murphy EB, Wudl F (2010) The world of smart healable materials. Prog Polym Sci 35:223–251
Kloxin CJ, Scott TF, Adzima BJ, Bowman CN (2010) Covalent Adaptable Networks (CANs): a unique paradigm in cross-linked polymers. Macromolecules 43:2643–2653
Binder WH (2013) Self-healing polymers: from principles to applications. Wiley-VCH, Weinheim
Blaiszik BJ, Kramer SLB, Olugebefola SC, Moore JS, Sottos NR, White SR (2010) Self-healing polymers and composites. Annu Rev Mater Res 40:179–211
Hager MD, Greil P, Leyens C, van der Zwaag S, Schubert US (2010) Self-healing materials. Adv Mater 22:5424–5430
Billiet S, Hillewaere XKD, Teixeira RFA, Du Prez FE (2013) Chemistry of crosslinking processes for self-healing polymers. Macromol Rapid Commun 34:290–309
White SR, Sottos NR, Geubelle PH, Moore JS, Kessler MR, Sriram SR, Brown EN, Viswanathan S (2001) Autonomic healing of polymer composites. Nature 409:794–797
Neuser S, Michaud V, White SR (2012) Improving solvent-based self-healing materials through shape memory alloys. Polymer 53:370–378
Fickert J, Makowski M, Kappl M, Landfester K, Crespy D (2012) Efficient encapsulation of self-healing agents in polymer nanocontainers functionalized by orthogonal reactions. Macromolecules 45:6324–6332
Chuo TW, Wei TC, Liu YL (2013) Electrically driven self-healing polymers based on reversible guest–host complexation of β-cyclodextrin and ferrocene. J Polym Sci Part A Polym Chem 51:3395–3403
Sheridan RJ, Bowman CN (2013) Understanding the process of healing of thermoreversible covalent adaptable networks. Polym Chem 4:4974–4979
Chen YL, Kushner AM, Williams GA, Guan ZB (2012) Multiphase design of autonomic self-healing thermoplastic elastomers. Nat Chem 4:467–472
Lu YX, Guan ZB (2012) Olefin metathesis for effective polymer healing via dynamic exchange of strong carbon–carbon double bonds. J Am Chem Soc 134:14226–14231
Deng GH, Li FY, Yu HX, Liu FY, Liu CY, Sun WX, Jiang HF, Chen YM (2012) Dynamic hydrogels with an environmental adaptive self-healing ability and dual responsive sol–gel transitions. ACS Macro Lett 1:275–279
Ying HZ, Zhang YF, Cheng JJ (2014) Dynamic urea bond for the design of reversible and self-healing polymers. Nat Commun. doi:10.1038/ncomms4218
Ling J, Rong MZ, Zhang MQ (2011) Coumarin imparts repeated photochemical remendability to polyurethane. J Mater Chem 21:18373–18380
Froimowicz P, Frey H, Landfester K (2011) You have full text access to this content towards the generation of self-healing materials by means of a reversible photo-induced approach. Macromol Rapid Commun 32:468–473
Chen XX, Dam MA, Ono K, Mal A, Shen HB, Nutt SR, Sheran K, Wudl F (2002) A thermally Re-mendable cross-linked polymeric material. Science 295:1698–1702
Scheltjens G, Diaz MM, Brancart J, Van Assche G, Van Mele B (2013) A self-healing polymer network based on reversible covalent bonding. React Funct Polym 73:413–420
Tian Q, Yuan YC, Rong MZ, Zhang MQ (2009) A thermally remendable epoxy resin. J Mater Chem 19:1289–1296
Zhang YC, Broekhuis AA, Picchioni F (2009) Thermally self-healing polymeric materials: the next step to recycling thermoset polymers. Macromolecules 42:1906–1912
Yang B, Zhang YL, Zhang XY, Tao L, Li SX, Wei Y (2012) Facilely prepared inexpensive and biocompatible self-healing hydrogel: a new injectable cell therapy carrier. Polym Chem 3:3235–3238
Ono T, Noboriab T, Lehn JM (2005) Dynamic polymer blends—component recombination between neat dynamic covalent polymers at room temperature. Chem Commun 1522–1524
Kamada J, Koynov K, Corten C, Juhari A, Yoon JA, Urban MW, Balazs AC, Matyjaszewski K (2010) Redox responsive behavior of thiol/disulfide-functionalized star polymers synthesized via atom transfer radical polymerization. Macromolecules 43:4133–4139
Canadell J, Goossens H, Klumperman B (2011) Self-healing materials based on disulfide links. Macromolecules 44:2536–2541
Lafont U, van Zeijl H, van der Zwaag S (2012) Influence of cross-linkers on the cohesive and adhesive self-healing ability of polysulfide-based thermosets. ACS Appl Mater Interfaces 4:6280–6288
Yoon JA, Kamada J, Koynov K, Mohin J, Nicolaÿ R, Zhang YZ, Balazs AC, Kowalewski T, Matyjaszewski K (2012) Self-healing polymer films based on thiol–disulfide exchange reactions and self-healing kinetics measured using atomic force microscopy. Macromolecules 45:142–149
Zhang YL, Tao L, Li SX, Wei Y (2011) Synthesis of multiresponsive and dynamic chitosan-based hydrogels for controlled release of bioactive molecules. Biomacromolecules 12:2894–2901
Xu ZY, Peng JX, Yan N, Yu H, Zhang SS, Liu KQ, Fang Y (2013) Simple design but marvelous performances: molecular gels of superior strength and self-healing properties. Soft Matter 9:1091–1099
Nakahata M, Takashima Y, Yamaguchi H, Harada A (2011) Redox-responsive self-healing materials formed from host–guest polymers. Nat Commun 2:511. doi:10.1038/ncomms1521
Cui JX, del Campo A (2012) Multivalent H-bonds for self-healing hydrogels. Chem Commun 48:9302–9304
Burattini S, Greenland BW, Merino DH, Weng WG, Seppala J, Colquhoun HM, Hayes W, Mackay ME, Hamley IW, Rowan SJ (2010) A healable supramolecular polymer blend based on aromatic π-π stacking and hydrogen-bonding interactions. J Am Chem Soc 132:12051–12058
Burattini S, Colquhoun HM, Fox JD, Friedmann D, Greenland BW, Harris PJF, Hayes W, Mackay ME, Rowan SJ (2009) A Self-repairing, supramolecular polymer system: healability as a consequence of donor–acceptor π-π stacking interactions. Chem Commun 6717–6719
Burnworth M, Tang LM, Kumpfer JR, Duncan AJ, Beyer FL, Fiore GL, Rowan SJ, Weder C (2011) Optically healable supramolecular polymers. Nature 472:334–338
Fox J, Wie JJ, Greenland BW, Burattini S, Hayes W, Colquhoun HM, Mackay ME, Rowan SJ (2012) High-strength, healable, supramolecular polymer nanocomposites. J Am Chem Soc 134:5362–5368
Smith AE, Xu XW, McCormick CL (2010) Stimuli-responsive amphiphilic (co) polymers via RAFT polymerization. Prog Polym Sci 35:45–93
Cheng CJ, Bai XX, Liu SJ, Huang QH, Tu YM, Wu HM, Wang XJ (2013) UV cured polymer based on a renewable cardanol derived RAFT agent. J Polym Res 20:197
Ma JY, Zhang HX (2014) Preparation and characterization of poly(methyl methacrylate)/SiO2 organic–inorganic hybrid materials via RAFT-mediated miniemulsion Polymerization. J Polym Res 21:590
Pan GQ, Zhang Y, Ma Y, Li CX, Zhang HQ (2011) Efficient One‐Pot synthesis of water‐compatible molecularly imprinted polymer microspheres by facile RAFT precipitation polymerization. Angew Chem Int Ed 50:11731–11734
Wang ZX, Zhang QH, Zhan XL, Chen FQ, Rao GH, Xiong JH (2013) Preparation, kinetics and microstructures of well-defined PS-b-PS/Bd diblock copolymers via RAFT miniemulsion polymerization. J Polym Res 20:288
Ganjeh-Anzabi P, Haddadi-Asl V, Salami-Kalajahi M, Abdollahi M (2013) Kinetic investigation of the reversible addition-fragmentation chain transfer polymerization of 1,3-butadiene. J Polym Res 20:248
Aoyagi N, Endo T (2009) Functional RAFT agents for radical-controlled polymerization: quantitative synthesis of trithiocarbonates containing functional groups as RAFT agents using equivalent amount of CS2. J Polym Sci A Polym Chem 47:3702–3709
Kim JS, Youk JH (2009) Preparation of core cross-linked micelles using a photo-cross-linking agent. Polymer 50:2204–2208
Amamoto Y, Kamada J, Otsuka H, Takahara A, Matyjaszewski K (2011) Repeatable photoinduced self-healing of covalently cross-linked polymers through reshuffling of trithiocarbonate units. Angew Chem Int Ed 50:1660–1663
Acknowledgments
The work was financially supported by Natural Science Foundations of China (NO. 21264008) and the Natural Science Foundations of Jiangxi Province (No. 2009GZH0035).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Cheng, C., Bai, X., Zhang, X. et al. Self-healing polymers based on a photo-active reversible addition-fragmentation chain transfer (RAFT) agent. J Polym Res 22, 46 (2015). https://doi.org/10.1007/s10965-015-0691-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-015-0691-9