
Abstract
In this study, novel polyimide/α-Fe2O3 (PI/α-Fe2O3) hybrid nanocomposite films were synthesized via thermal curing of different amount of modified α-Fe2O3 nanoparticles containing poly(amic acid) derived from 3,5-diamino-N-(9H-fluoren-2-yl)benzamide and 3,3′,4,4′-benzophenonetetracarboxylic dianhydride in N-methyl-2-pyrrolidone. To improve the dispersion and obtain the homogeneous distribution of α-Fe2O3 in polymeric matrix, the surface of nanoparticles was modified to organophile with γ-aminopropyltriethoxy silane. The PI/α-Fe2O3 nanocomposite films were characterized by Fourier transform infrared spectra, X-ray diffraction and transmission electron microscopy (TEM). The TEM results confirm that the nanoparticles were dispersed uniformly in PI matrix on nanoscale. Additionally, thermogravimetric analysis data showed an improvement of thermal stability of novel nanocomposite films as compared to the pure polymer.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Organic/inorganic hybrid materials have become a popular topic in material science due to their unique mechanical, electrical, magnetic and adhesive properties [1–9]. Polyimides (PI)s are accepted as reliable high-temperature polymers. They are widely utilized in microelectronic devices because of their excellent thermomechanical and planarization properties, low dielectric constants, and high dissipation factors [10–19]. Therefore, PIs are a promising matrix candidate for hybrid materials [20–22].
Nanostructured hematite (α-Fe2O3) is a typical environmentally friendly semiconductor material. Nanostructured α-Fe2O3 is the most thermodynamically stable iron oxide phase and is of particular interest owing to its high resistance to corrosion, low processing cost and non-toxicity [23]. Therefore, this multifunctional material has investigated extensively for a variety of applications including photo-catalysis [24–26], gas sensing [27–30], photo-assisted electrolysis of water [31, 32], magnetic recording [29, 33], drug delivery [34], tissue repair engineering [35] and magnetic resonance imaging [36], along with its use in lithium-ion batteries [29], spin electronic devices [37] and pigments [38]. In particular, the magnetic properties of α-Fe2O3 have attracted much interest over the past decades. In the 1950s it was found to be canted antiferromagnetic (weakly ferromagnetic) at room temperature, antiferromagnetic below the Morin transition temperature of 250 K and paramagnetic above its Neel temperature of 948 K.
In the present article, we report the synthesis and characterization of PI/α-Fe2O3 nanocomposite films using amine-functionalized Fe2O3 and poly(amic acid) (PAA) derived from 3,5-diamino-N-(9H-fluoren-2-yl)benzamide and 3,3′,4,4′-benzophenonetetracarboxylic dianhydride as the PI precursor. The resulting nanocomposites were characterized using Fourier transform infrared (FT-IR), thermogravimetric analysis (TGA), X-ray diffraction (XRD), and transmission electron microscopy (TEM).
Experimental
Materials
All chemicals were purchased from Fluka Chemical Co. (Buchs, Switzerland), Aldrich Chemical Co. (Milwaukee, WI), Riedel-deHaen AG (Seelze, Germany) and Merck Chemical Co. 2-Aminofluorene, hydrazine monohydrate, 10 % palladium on activated carbon (Pd/C) and 3,5-dinitrobenzoyl chloride (from Merck) were used as received. 3,3′,4,4′-Benzophenonetetracarboxylic dianhydride (BTDA, from Merck) and 4,4′-(hexaflouroisopropylidene)diphthalic anhydride (6FDA, from Aldrich) were recrystallized from acetic anhydride before use. Pyromellitic dianhydride (PMDA, from Merck) and naphthalene-1,4,5,8-tetracarboxylic dianhydride (NTDA, from Aldrich) were purified by sublimation. N-Methyl-2-pyrrolidinone (NMP) was purified by distillation under reduced pressure over barium oxide. α-Fe2O3 nanoparticles were purchased from Neutrino Co. The silane coupling agent (γ-aminopropyltriethoxy silane) (APTES, KH550) was purchased from Merck Chemical Co.
Techniques
Proton nuclear magnetic resonance (1H NMR) spectra were recorded on Bruker Avance 400 MHz spectrometer in dimethyl sulfoxide-d6 (DMSO-d6). The proton resonances were designated as singlet (s) and multiplet (m). Inherent viscosities were measured by a standard procedure using a Cannon-Fenske routine viscometer (Germany) at the concentration of 0.5 g/dL at 25 °C. FT-IR spectra were recorded with a Jasco-680 spectrometer (Japan) in the range of 400–4000 cm−1. Vibration bands were reported as wavenumber (cm−1). FT-IR spectra of all samples were collected by making their pellets in KBr as a medium. The diffraction pattern of related materials was recorded in the reflection mode using a Bruker, D8 Advance diffractometer. Nickel filtered CuKα radiation (radiation wavelength, λ =0.154 nm) was produced at an operating voltage of 45 kV and a current of 100 mA. The redox behavior was investigated with cyclic voltammetry on a potentiostat EG&G Co, PARSTAT 2273, USA instrument. It was conducted for the dipped polymer film on the working electrode in dry acetonitrile containing tetrabutylammonium hexafluorophosphate (0.1 M) as an electrolyte. Surface morphology and sample homogeneity of polymer nanocomposites were characterized using FE-SEM HITACHI (S-4160) (Tokyo, Japan). The morphology and dispersity analysis were performed on transmission electron micrograph (TEM) analyzer on Philips CM 120 operating at 100 kV. The reaction was carried out on a Misonix ultrasonic liquid processor, XL-2000 Series. Ultrasound was a wave of frequency 2.25 × 104 Hz and power 100 W. Thermogravimetric analysis (TGA) is performed with a STA503 win TA at a heating rate of 10 °C/min from 25 °C to 800 °C under nitrogen atmosphere.
Monomer synthesis
Synthesis of N-(9H-fluoren-2-yl)-3,5-dinitrobenzamide (3)
Into a 25 mL round-bottomed flask fitted with a magnetic stirrer was placed a solution of 0.200 g (1.10 × 10−3 mol) 2-aminofluorene (1) in 3 mL of dry NMP. The reaction mixture was cooled in an ice water bath. To this solution 0.254 g (1.10 × 10−3 mol) of 3,5-dinitrobenzoyl chloride (2) in 2 mL of NMP was added dropwise. The mixture was stirred in ice bath for 3 h, and then 0.75 mL of triethylamine was added. The mixture was stirred in ice bath for 2 h and at room temperature overnight. The resulting mixture was poured into 10 mL/30 mL cold water/concentrated HCl. The precipitate was collected by filtration and washed thoroughly with water and dried at 70 °C for 8 h to yield 0.39 g (95 %) of compound 3. Recrystallization from DMF/water gave orange crystals. mp 309 °C. FT-IR (KBr, cm−1): 3262 (N-H stretch), 1651 (C = O stretch), 1538, 1341 (−NO2 stretch). 1H NMR (DMSO-d 6, ppm): 10.94 (s, 1H, N-H), 9.21 (d, 2H, Ar, J =7.2 Hz), 9.01 (t, 1H, Ar, J =2.1 Hz), 8.13 (d, 1H, Ar, J =0.8 Hz), 7.93 (d, 1H, Ar, J =8.3 Hz), 7.87 (d, 1H, Ar, J =7.5 Hz), 7.79 (dd, 1H, Ar, J =1.8 Hz, J =8.3 Hz), 7.59 (d, 1H, Ar, J =7.4 Hz), 7.40 (t, 1H, Ar, J =7.2 Hz), 7.31 (dt, 1H, Ar, J =1.0 Hz, J =7.4 Hz), 3.98 (s, 2H, CH2).
Synthesis of 3,5-diamino-N-(9H-fluoren-2-yl)benzamide (4)
0.200 g (5.33 × 10−4 mol) of the dinitro compound 3, 0.01 g of 10 % Pd/C, and 10 mL of ethanol were introduced into a two-necked flask to which 3 mL of hydrazine monohydrate was added dropwise at 60 °C. After the complete addition, the reaction was continued at reflex temperature for another 12 h. To the suspension, 10 mL of THF was added to redissolve the precipitated product, and refluxing was continued for 90 min. The mixture was filtered to remove the Pd/C and the filtrate was distilled to remove the solvent. The crude product recrystallized from ethanol and dried in vacuo at 80 °C to give 0.14 g (yield: 86 %) of compound 4. mp 255 °C. FT-IR (KBr, cm−1): 3466, 3439, 3360 and 3330 (N-H stretch), 1650 (C = O stretch). 1H NMR (DMSO-d 6, ppm): 10.03 (s, 1H, N-H), 8.09 (d, 2H, Ar, J =1.1 Hz), 7.83 (d, 2H, Ar, J =8.3 Hz), 7.71 (dd, 1H, Ar, 1 J =1.9 Hz, 2 J = 8.3), 7.56 (d, 1H, Ar, J =1.9 Hz), 7.36 (t, 1H, Ar, J =6.8 Hz), 7.27 (dt, 1H, Ar, 1 J =1.1 Hz, 2 J =7.4 Hz), 6.31 (d, 2H, Ar, J =1.9 Hz), 6.01 (t, 1H, Ar, J =1.9 Hz), 4.96 (s, 4H, 2NH2), 3.91 (s, 2H, CH2).
Polymer synthesis
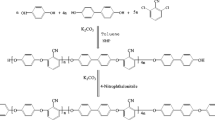
The typical two-step method was applied to the polycondensation of monomers. 0.2043 g (6.34 × 10−4 mol) of BTDA was added gradually to a solution consisting of 0.2000 g (6.34 × 10−4 mol) diamine 4 dissolving in 3 mL of NMP with stirring. Thus the solid content of the mixture solution was approximately 10 %. The mixture was stirred at room temperature for 24 h under nitrogen atmosphere to yield a viscous PAA solution in NMP. The inherent viscosity of the PAA in DMAc was 0.62 dL/g, measured at a concentration of 0.50 g/dL at 25 °C. The PAA was subsequently converted into PI by either a thermal or chemical imidization process. For the thermal imidization, the PAA film was obtained by casting from the reaction polymer solution onto a glass plate and drying at 90 °C overnight under vacuum. Then the PAA film was transformed into PI by sequentially heated at 110, 150, 180, 210, 230 and 280 °C for 30 min each. Meanwhile, chemical imidization was carried out via the addition of 3 mL of an acetic anhydride/pyridine (5:4, v/v) mixture into the PAA solution at ambient temperature, then the mixture was stirring at 100 °C for 3 h to yield a homogeneous PI solution, which was poured slowly into methanol to give a fibrous precipitate, which was collected by filtration, washed thoroughly with methanol, dried at 100 °C in vacuum.
PAA: FT-IR (KBr, cm−1): υ =3400-2400 (br), 1781 (m), 1724 (s), 1668 (s), 1594 (s), 1536 (s), 1488 (m), 1454 (m), 1290 (m), 1091 (m), 987 (m), 863 (m), 796 (m), 765 (m), 649 (m).
PI-b-I :FT-IR (KBr, cm−1): 3385 (N-H stretch), 1785 (asymmetrical C = O, imide), 1729 (symmetrical C = O, imide), 1670 (C = O stretch, amide), 1351 (C-N stretch), 720 (imide ring deformation).
PI-a-II :1H NMR (DMSO-d 6, ppm): 11.15 (s, 1H, N-H), 8.63–6.82 (m, Ar), 5.76 (s, 1H, CH), 4.03 (s, 1H, CH).
PI-c-II :1H NMR (DMSO-d 6, ppm): 11.00 (s, 1H, N-H), 8.55–6.89 (m, Ar), 5.76 (s, 1H, CH), 4.04 (s, 1H, CH).
Surface functionalization of α-Fe2O3 nanoparticles with APTES
α-Fe2O3 nanoparticles were dried at 110 °C in an oven for 24 h to remove the adsorbed water. Modification of α-Fe2O3 nanoparticles with γ-aminopropyltriethoxy silane (APTES, H2N(CH2)3Si(OCH2CH3)3) was carried out as follows. A silane-coupling agent, APTES was added to a toluene solution containing α-Fe2O3 nanoparticles, which had been vigorously stirred overnight to disperse 0.1 g of α-Fe2O3 nanoparticles homogeneously in 30 mL of toluene. A further day was spent for introducing APTES moieties to the surface of α-Fe2O3 nanoparticles. After filtering and washing by copious amounts of toluene and acetone in sequence, the final product was attained as a bright brown powder.
Preparation of PI/α-Fe2O3 nanocomposites
PI/α-Fe2O3 nanocomposites were prepared with different weight percentages of α-Fe2O3 (5, 10, 15 and 20) as follow:
0.2000 g (6.34 × 10−4 mol) diamine 4 and 0.2043 g (6.34 × 10−4 mol) of BTDA in 5 mL of NMP gave viscous gel of PAA, which was applied for the experiment. Different weight percentages of α-Fe2O3 (5, 10, 15 and 20 wt %) were added to the PAA, and the suspension was sonicated for 4.5 h. The nanocomposite films were then cast from the suspension placed onto the dust-free glass plates. After the film production, they were annealed at 80, 100, 200, and 300 °C for 1 h each to convert the PAA into PI.
PI/α-Fe2O3 10 wt.%: FT-IR (KBr, cm−1): υ =3419 (m), 1779 (m), 1722 (s), 1670 (s), 1590 (s), 1533 (s), 1488 (s), 1454 (s), 1421 (s), 1390 (s), 1348 (s), 1294 (s), 1234 (s), 1191 (s), 1159 (s), 1089 (s), 983 (m), 892 (m), 854 (m), 769 (m), 715 (s), 622 (m), 545 (s), 472 (m), 422 (m).
Results and discussion
Monomer synthesis
N-(9H-Fluoren-2-yl)-3,5-dinitrobenzamide (3) was prepared by the condensation of 2-aminofluorene (1) and 3,5-dinitrobenzoyl chloride (2). The new aromatic diamine having a bulky pendent fluorenamide group, 3,5-diamino-N-(9H-fluoren-2-yl)benzamide (4), was successfully synthesized by hydrazine Pd/C-catalytic reduction according to the synthetic route outlined in Scheme 1. The purity of monomer 4 was checked by thin layer chromatography, which showed one spot in an ethylacetate/cyclohexane mixture (50:50) with Rf =0.38. FT-IR and 1H NMR spectroscopic techniques were applied to identify the structure of the compound 3 and the diamine monomer 4. The transformation of nitro to amino functionality could be monitored by the change of FT-IR spectra. The nitro groups of compound 3 gave two characteristic bands at around 1538 and 1341 cm−1 (−NO2 asymmetric and symmetric stretching). After reduction, the characteristic absorptions of the nitro group disappeared and the amino group showed the typical N-H stretching absorption pair in the region of 3466–3330 cm−1. The 1H NMR spectra of compound 3 and the diamine monomer 4 are shown in Figs. 1 and 2. These spectra agree well with the proposed molecular structure of compounds 3 and 4. The 1H NMR spectrum confirm that the nitro groups have been completely transformed into amino groups by the high-field shift of the aromatic protons and by the resonance signals at 4.96 ppm corresponding to the amino protons.

Synthesis of diamine 4 and PIs

1H NMR spectrum of dinitro 3 in DMSO-d6 solution

1H NMR spectrum of diamine 4 in DMSO-d6 solution
Polymer synthesis
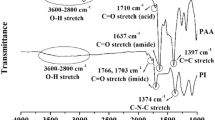
The new PIs having fluorenamide units were prepared by the polycondensation of equal molar amounts diamine monomer 4 with various aromatic dianhydride monomers through the conventional two-step imidization technique, which involved ring opening polyaddition of monomers to form PAAs and subsequently cyclodehydration to PIs, as shown in Scheme 1. The PAA solution was prepared by slowly adding a solution of dianhydride to a solution of the diamine 4 in anhydrous NMP. The either thermal or chemical imidization procedures were chosen to attain imidization at the final stage of the reaction forming PIs, merits of the former were ease for the synthesis, while the latter was suited for the preparation of soluble PI. The chemical cyclodehydration of the PAA was made in the presence of a mixture of acetic anhydride and pyridine as dehydrating agents. The chemical conversion also enabled us to more easily characterize the resulting PIs. Alternatively, the thermal cyclodehydration of the PAA was occurred via heating the PAA films to the temperature 280 °C under vacuum to fabricate PI films. The results of the polymerization reactions are presented in Table 1. Inherent viscosity values of PIs were in the range of 0.33–0.44 dL/g showing a moderate molecular weight. These polymers generally exhibited brown color due to charge transfer between the diamine donor moieties and dianhydride acceptor groups (Table 1). The structure of the PIs was confirmed by FT-IR and 1H NMR spectroscopic analyses. The absorptions at around 3500–2700 cm−1 and 1700–1600 cm−1 presented in FT-IR spectrum of PAA should be assigned to the -OH vibration of carboxyl bands and the carbonyl group vibration of amide bands in PAA (Fig. 9d), and they disappear after thermal or chemical imidization, however, there exist strong absorptions that represent imide ring at 1785 and 1729 cm−1 after forming PIs. Comparing with FT-IR spectra of PIs, there exist similar IR absorption bands for the PI either from thermal or chemical imidization, these show that the conversion from PAA to PI were basically complete using two kinds of imidization methods. Figures 3 and 4 show the high-resolution 1H NMR spectra of PI-a-I and PI-c-I by chemical imidization, respectively. The assignments of the 1H NMR spectra agree well with the proposed polymer structure. The above results reveal that the diamine monomer 4 holds a good polymerization activity to form PIs, meanwhile, 100 % chemical imidization could be achieved at lower temperatures, and this should be suitable to get soluble PI.

1H NMR spectrum of PI-a-I in DMSO-d6 solution

1H NMR spectrum of PI-c-I in DMSO-d6 solution
The solubility of PIs was tested quantitatively in various solvents. Most of the PIs obtained by chemical imidization, were soluble in the test solvents including NMP, DMAc, DMF and DMSO and were insoluble in solvents such as chloroform, methylene chloride, acetone, methanol, ethanol and water. The solubility of the PIs in several organic solvents is also summarized in Table 2. The good solubility should be result from increased flexibility or free volume caused by the introduction of a bulky pendent fluorenamide group in the repeat unit. Comparing with those obtained by chemical imidization, the poor solubility of PIs obtained by thermal imidization was possibly due to the presence of partial inter molecular crosslinking during the thermal imidization stage. The excellent solubility makes these PIs potential candidates for practical applications in spin- or dip-coating processes.
The UV–vis spectra of the PIs were recorded in DMF. It is apparent that the wavelength of maximum absorption is related to the π → π* transition resulting from the conjugation between the aromatic rings and nitrogen atoms in the aforementioned compounds. All of these compounds show almost similar UV–vis spectra pattern. These polymers showed maximum absorption at 318–357 nm in DMF solution. The absorption spectrum of the PI-c-II is shown in Fig. 5.

Absorption spectrum of PI-c-II in DMF solution 2.70 × 10−5 M
Charge transport in organic materials is believed to be governed by the hopping process involving redox reaction of charge transport molecules. Cyclic voltammetry (CV) is a preliminary characterization method to determine the redox properties of polymeric materials. Figure 6 shows the representative cyclic voltammogram of polymer PI-a-I in acetonitrile with 0.1 M Bu4NPF6. One pair of redox waves was observed in these polymers. As shown in this figure, PI-a-I showed an oxidation wave of which a peak top is at 1.3 V (versus Ag/AgCl).

Cyclic voltammogram of PI-a-I in acetonitrile with 0.1 M Bu4NPF6
Surface modification of α-Fe2O3 nanoparticles
Surface-treating α-Fe2O3 filler with a suitable modifying agent can improve the compatibility of α-Fe2O3 filler with polymeric matrix, promotes effectively the adhesion of polymeric matrix to α-Fe2O3 filler; and reduces the aggregation of nanoparticles in polymer/α-Fe2O3 nanocomposite. In this investigation, the surface of α-Fe2O3 nanoparticles was treated via APTES. Hydroxyl groups that exist on the surface of α-Fe2O3 nanoparticles can replace with OCH2CH3 of the APTES to bond to it (Fig. 7). In this way, the surface of α-Fe2O3 was changed to hydrophobic that can have good affinity to organic matrix. As a result, the modified α-Fe2O3 nanoparticles can fulfill steric hindrance between inorganic nanoparticles and organic polymer to obtain uniform dispersion and prevent the aggregation process. For coupling agent APTES, the functional group that provides different interactions to α-Fe2O3 nanoparticle is the amino group. Various interaction types between aminosilane and α-Fe2O3 surface were proposed as follows: a) hydrogen bonding b) ionic bonding c) covalent bonding with surface hydroxyl groups. The details of the mechanism are shown in Fig. 8. FT-IR spectra of as-received α-Fe2O3 nanoparticles and APTES-modified α-Fe2O3 nanoparticles are shown in Fig. 9 together with the spectrum of pure APTES molecule. A broad absorption band in the range from 900 to 1100 cm−1 observed in the FT-IR spectrum of the APTES-modified α-Fe2O3 nanoparticles (c) is assignable to the Si-O stretching. The disappearance of the peaks at 1170, 1110, 1080 and 960 cm−1, which are characteristic bands of Si-O-C in APTES observed in spectrum of pure APTES (b) and the appearance of the bands at 1130 and 1010 cm−1, not observed in spectrum of pure α-Fe2O3 nanoparticles (a), are indicative of the formation of Si-O-Fe and Si-O-Si bonds resulting from the reaction of APTES molecules. In addition, the enhancement of the absorption band in the range from 1450 to 1600 cm−1, compared to spectrum of pure α-Fe2O3 nanoparticles (a) collected for the pre-modified sample, is attributed to the overlap of the CH2 bending and the N-H bending originated from the introduction of APTES moieties. Furthermore, small peaks at about 2940 and 2850 cm−1, attributable to asymmetric and symmetric CH2 stretching, respectively, appear in spectrum of modified α-Fe2O3 nanoparticles (c), whereas the peaks at 2960 and 2880 cm−1 in spectrum of pure APTES (b) assigned to the asymmetric and symmetric CH3 stretching, respectively, in the ethoxy group of APTES are not observed in spectrum of modified α-Fe2O3 nanoparticles (c). These results suggest that the silane-coupling reaction progressed at the surface of nanoparticles leading to the chemical binding of the APTES group to the surface of α-Fe2O3 nanoparticles [39].

Surface modification of α-Fe2O3 nanoparticles with APTES

The mechanism of PI/α-Fe2O3 formation

FT-IR spectra of: (a) as-recevied α-Fe2O3 nanoparticles; (b) APTES-modified α-Fe2O3 nanoparticles; (c) pure APTES; (d) PAA; (e) PI/α-Fe2O3 5 wt%
Nanocomposite synthesis
The polymerization of diamine 4 and BTDA in dry NMP proceeds at the room temperature to avoid the premature imidization of amic acid moieties. Obtained PAA was mixed with modified α-Fe2O3 nanoparticles and the solution was sonicated for 4.5 h. The thin films of the stated crude mixtures were prepared by casting on dust-free glass plates. Then gradual annealing of thin films was performed up to 300 °C and continued at this temperature for several hours to ensure the complete imidization.
The FT-IR spectrum of the PI/α-Fe2O3 nanocomposite 5 wt% shown in Fig. 9e has all the frequencies of pure PI with additional frequencies around 450 and 550 cm −1, which correspond to the Fe-O vibration frequencies of pure α-Fe2O3. There was a slight blue shift in these two peaks when compared with the pure α-Fe2O3. This blue shift observed in the FT-IR spectra of the nanocomposites was possibly because of the weak bonding formed between PI and α-Fe2O3.
XRD analysis of the nanocomposite films was performed to determine the chemical state of α-Fe2O3 incorporated within PI film. XRD patterns of the pure α-Fe2O3 nanoparticles (a), PI/α-Fe2O3 nanocomposite 5 wt% (b) and PI-b-I (c) are given in Fig. 10. The peaks of α-Fe2O3 nanoparticles in the range of 2θ =20–80° correspond to (021), (104), (110), (113), (024), (116), (018), (214), (300), (010) and (217) Bragg reflection, respectively, and identify that α-Fe2O3 nanoparticles have a rhombohedral structure. The full width at half-maximum of the strongest characteristic reflection (104) is used to estimate the average crystallite size by applying the Scherrer equation. The crystallite size of α-Fe2O3 nanoparticles in PI film is 50 nm, which is consistent with the result determined by statistical analysis of the TEM image, indicating that each individual particle is a single crystal. The diffraction pattern for the pure PI shown in Fig. 10c demonstrates that all the PIs are completely amorphous, primarily because of the bulky fluorenamide pendant groups. Furthermore, the amorphous nature of these polymers is also reflected in their good solubility.

X-ray diffraction patterns of (a) Pure α-Fe2O3 nanoparticles; (b) PI/α-Fe2O3 nanocomposite 5 wt% and (c) PI-b-I
According to the electronic structure of Fe2O3 reported previously [40], the optical absorption band in the visible and UV regions were attributed to the transition in the crystal field and the charge-transfer processes, respectively. The polymer has absorption about 343 nm. In the case of the nanocomposites, the strongest absorption band is assigned to the electron transfer from O 2p to Fe 3d, corresponding to the valance band to the conductive band transition, and the small shoulder at 480 nm is considered to arise from a crystal-field spin-forbidden transition (d-d transition).
To characterize the morphology and dispersity α-Fe2O3 nanoparticles in the nanocomposites, FE-SEM images of fracture surfaces of neat PI and nanocomposite 15 wt.% were examined. The neat PI showed a smooth fracture surface morphology. For PI/α-Fe2O3 nanocomposite, α-Fe2O3 nanoparticles were well dispersed and embedded in the PI matrix without showing noticeable α-Fe2O3 aggregates. In addition, the fracture surface of the PI/α-Fe2O3 nanocomposite is relatively smooth without exhibiting the pull-out of α-Fe2O3 nanoparticles from the PI matrix and the boundary between modified α-Fe2O3 and PI matrix is not discernibly clear. These results strongly support that due to hydrogen bonding between polar and hydrophilic groups of modified α-Fe2O3 and polar groups in the PI, the α-Fe2O3 nanoparticles are wrapped around PI particles and nanoparticles are also mixed well with the polymer matrix, eventually leading to the good dispersity of α-Fe2O3 nanoparticles in the PI matrix. Silane treatment of inorganic nanoparticles is a usual and effective way to make a strong connection between a polymer and nanoparticles, because of the unique bifunctional structure of the silane coupling agents with one end being capable of reacting with the hydroxyl groups on the nanoparticles surface and the other end being compatible with the polymer. The modification process is described as a hydrolysis and condensation reaction between the silane coupling agents and the nanoparticles surface in a polar medium. The bonding between the silane and the nanoparticles surface removes the surface hydroxyl groups and changes the hydrophilic surface into a hydrophobic surface. So the result of surface treatment is a reduction of the nanoparticles-nanoparticles interaction and an improvement in the dispersability of the nanoparticles in the polymer matrix. Therefore, the aggregation of nano-α-Fe2O3 in the PI matrix decreased after it was modified by the silane coupling agent KH550.
TEM analysis was performed for PI/α-Fe2O3 nanocomposite films to identify the distribution of nanoparticles within polymer matrix. Figure 11 shows the TEM image of the nanocomposite 15 wt.%. As can be seen, the nanoparticles were well dispersed in the polymer matrix. The relatively strong interactions between polymer matrix and α-Fe2O3 nanoparticles are responsible for observing the nanoparticles with almost spherical shapes.

TEM image of nanocomposite 10 %
The thermal properties of pure PIs and PI hybrid nanocomposite 15 wt.% were studied using TGA at a heating rate of 10 °C min−1, under nitrogen atmosphere. The TGA curves of pure PI and PI hybrid nanocomposites 15 wt.% were shown in Figs. 12 and 13. The thermoanalysis data of these samples are summarized in Table 3. These studies show that the nanocomposites are thermally stable up to 500 °C. The 10 % weight loss temperatures of the pure PI and nanocomposite were recorded in 526 and 585 °C for PI-b-II and PI-b-II/α-Fe2O3 15 wt% respectively. The amount of residue (char yield) of nanocomposit, PI-b/α-Fe2O3 15 wt% was more than 63 % at 800 °C. As shown in the Figs. 12 and 13, the thermal stability of nanocomposite is higher than pure PI that is due to the good compatibility of modified α-Fe2O3 particles with polymer matrix. α-Fe2O3 nanoparticles have high thermal stability due to their larger surface area, so the incorporation of α-Fe2O3 nanoparticles can improve the thermal resistance of nanocomposites. The DSC thermogram of PI-b-II/α-Fe2O3 15 wt% showed high glass transition temperatures about 294 °C (Fig. 14). The presence of α-Fe2O3 nanoparticles in a polymer matrix improved the thermal stability and fire retardant properties of the polymer.

TGA thermogram of PI-b-II

TGA thermogram of PI-b/α-Fe2O3 15 wt%

DSC thermogram of PI-b-II/α-Fe2O3 15 wt%
Char yield can be applied as decisive factor for estimating limiting oxygen index (LOI) of the polymers based on Van Krevelen and Hoftyzer equation [41].
where CR = char yield.
The nanocomposite had LOI values calculated derived from their char yield was higher than 42. On the basis of LOI values, such nanocomposites can be classified as self-extinguishing materials.
Conclusions
A series of novel aromatic PIs with pendent fluorenamide moieties were prepared from 3,5-diamino-N-(9H-fluoren-2-yl)benzamide and various tetracarboxylic dianhydrides via the direct polycondensation. These polymers were thermally stable and soluble in common organic solvents. Novel PI/α-Fe2O3 nanocomposites were fabricated by ultrasonic technique as a simple and inexpensive route to prepare the polymer/nanoparticles, using α-Fe2O3 nanoparticles modified by APTES. The surface modification of inorganic particles by similar molecular chains to those of an organic matrix is a practical way to improve the compatibility between the organic and the inorganic phases. The modification would create the nanoparticles scatter within the matrix more homogeneously. Furthermore, the molecules grafted onto the nanoparticles would interact with the matrix, hence the final mechanical properties could be greatly improved. FT-IR and XRD data confirmed that α-Fe2O3 nanoparticles are dispersed in the PI matrix. Morphology investigation of resulting nanocomposite exhibited well-dispersed α-Fe2O3 nanoparticles in the polymer matrix by TEM analysis. TGA studies indicated that thermal stability of the nanocomposites has improved as compared to the pure polymer. This method offers a simple way for preparation PI/α-Fe2O3 nanocomposite films for potential application in high-density data-storage media.
References
Dasgupta D, Shishmanova IK, Ruiz-Carretero A, Lu K, Verhoeven M, van Kuringen HPC, Portale G, Leclère P, Bastiaansen CWM, Broer DJ, Schenning APHJ (2013) Patterned silver nanoparticles embedded in a nanoporous smectic liquid crystalline polymer network. J Am Chem Soc 135:10922–10925
Ignat M, Ovezea D, Hamciuc E, Hamciuc C, Dimitrov L (2014) Study on the electromechanical properties of polyimide composites containing TiO2 nanotubes and carbon nanotubes. J Polym Res 21:536–547
Zhang QG, Liu QL, Zhu AM, Xiong Y, Zhang XH (2008) Characterization and permeation performance of novel organic–inorganic hybrid membranes of poly(vinyl Alcohol)/1,2-bis(triethoxysilyl)ethane. J Phys Chem B 112:16559–16565
Naga N, Miyanaga T, Furukawa H (2010) Synthesis and optical properties of organic–inorganic hybrid gels containing fluorescent molecules. Polymer 51:5095–5099
Gofman I, Zhang B, Zang W, Zhang Y, Song G, Chen C, Li Y (2013) Specific features of creep and tribological behavior of polyimide-carbon nanotubes nanocomposite films: effect of the nanotubes functionalization. J Polym Res 20:258–266
Tsai MH, Liu SJ, Chiang PC (2006) Synthesis and characterization of polyimide/titania nano hybrid films. Thin Solid Films 515:1126–1131
Zhan J, Tian G, Jiang L, Wu Z, Wu D, Yang X, Jin R (2008) Superparamagnetic polyimide/γ-Fe2O3 nanocomposite films: preparation and characterization. Thin Solid Films 516:6315–6320
Jiang LY, Leu CM, Wei KH (2002) Layered silicates/fluorinated polyimide nanocomposites for advanced dielectric materials applications. Adv Mater 14:426–429
Wahab MA, Kim I, Ha CS (2004) Microstructure and properties of 3,3′,4,4′-biphenyltetracarboxylic dianhydride (BPDA)-p-phenylene diamine (PDA) polyimide/poly(vinylsilsesquioxane) hybrid nanocomposite films. J Polym Sci Part A: Polym Chem 42:5189–5199
Mittal KL (1984) Polyimides: synthesis, characterization, and applications. Plenum Press, New York
Ghosh MK, Mittal KL (1996) Polyimides: Fundamentals and applications. Dekker M (Ed.), New York
Bauer CL, Farris RJ (1989) In Polyimides: Materials, chemistry and characterization. Feger C, Khojasteh MM, McGrath JE. Eds.; Elsevier: Amsterdam
Gong S, Liu M, Xia S, Wang Y (2014) Synthesis of novel soluble polyimides containing triphenylamine groups for liquid crystal vertical alignment layers. J Polym Res 21:542–552
Hsiao SH, Yeh SJ, Wang HM, Guo W, Kung YR (2014) Synthesis and optoelectronic properties of polyimides with naphthyldiphenylamine chromophores. J Polym Res 21:407–418
Luo L, Pang Y, Jiang X, Wang X, Zhang P, Chen Y, Peng C, Liu X (2011) Preparation and characterization of novel polyimide films containing amide groups. J Polym Res 19:9783–9789
Moghanian H, Mobinikhaledi A, Baharangiz Z (2014) Synthesis, characterization and magnetic properties of novel heat resistant polyimide nanocomposites derived from 14H-dibenzo [a, j] xanthenes. J Polym Res 21:513–528
Yi L, Li C, Huang W, Yan D (2014) Soluble aromatic polyimides with high glass transition temperature from benzidine containing tert-butyl groups. J Polym Res 21:572–581
Huang X, Huang W, Fu L, Yan D (2012) Synthesis and characterization of thioether-containing polyimides with high refractive indices. J Polym Res 19:9790–9798
Chen Y, Zhang Q (2014) Synthesis and properties of polyimides derived from diamine monomer containing bi-benzimidazole unit. J Polym Res 21:424–431
Lin J, Liu Y, Yang W, Xie Z, Zhang P, Li X, Lin H, Chen G, Lei Q (2014) Structure and mechanical properties of the hybrid films of well dispersed SiO2 nanoparticle in polyimide (PI/SiO2) prepared by sol–gel process. J Polym Res 21:531–538
Park OK, Hwang JY, Goh M, Lee JH, Ku BC, You NH (2013) Mechanically strong and multifunctional polyimide nanocomposites using amimophenyl functionalized graphene nanosheets. Macromolecules 46:3505–3511
Akhter T, Saeed S, Siddiqi HM, Park OO, Ali G (2013) Synthesis and characterization of novel coatable polyimide-silica nanocomposites. J Polym Res 21:332–342
Cornell RM, Schwertman U (2003) The Iron Oxides, 2nd edn. Wiley-VCH Verlag Gmbh & Co, KGaA, Weinheim
Zheng Y, Cheng Y, Wang Y, Bao F, Zhou L, Wei X, Zhang Y, Zheng Q (2006) Quasicubic alpha-Fe2O3 nanoparticles with excellent catalytic performance. J Phys Chem B 110:3093–3097
Li LL, Chu Y, Liu Y, Dong LH (2007) Template-free synthesis and photocatalytic properties of novel Fe2O3 hollow spheres. J Phys Chem C 111:2123–2127
Pulgarin C, Kiwi J (1995) Iron oxide-mediated degradation, photodegradation, and biodegradation of aminophenols. Langmuir 11:519–526
Wu Z, Yu K, Zhang S, Xie Y (2008) Hematite hollow spheres with mesoporous shell: controlled synthesis and applications in gas sensor and lithium ion battery. J Phys Chem C 112:11307–11313
Hu X, Yu JC, Gong J, Li Q, Li G (2007) α-Fe2O3 Nanorings prepared by a microwave-assisted hydrothermal process and their sensing properties. Adv Mater 19:2324–2329
Wu C, Yin P, Zhu X, Yang CO, Xie Y (2006) Synthesis of hematite (α-Fe2O3) nanorods: diameter-size and shape effects on their applications in magnetism, lithium ion battery, and gas sensors. J Phys Chem B 110:17806–17812
Pelino M, Colella C, Cantallini C, Faccio M, Ferri G, D’Amico A (1992) Microstructure and electrical properties of an α-hematite ceramic humidity sensor. Sens Actuat B: Chem 7:464–469
Iida H, Nakanishi T, Osaka T (2005) Surface modification of γ-Fe2O3 nanoparticles with aminopropylsilyl groups and interparticle linkage with α, ω-dicarboxylic acids. Electrochim Acta 51:855–859
Zeng S, Tang K, Li T, Liang Z, Wang D, Wang Y, Zhou W (2007) Hematite hollow spindles and microspheres: Selective synthesis, growth mechanisms, and application in lithium ion battery and water treatment. J Phys Chem C 111:10217–10225
Sivula K, Zboril R, Formal FL, Robert R, Weidenkaff A, Tucek J, Frydrych J, Grätzel M (2010) Photoelectrochemical water splitting with mesoporous hematite prepared by a solution-based colloidal approach. J Am Chem Soc 132:7436–7444
Zeng S, Tang K, Li T, Liang Z, Wang D, Wang Y, Qi Y, Zhou W (2008) Facile route for the fabrication of porous hematite nanoflowers: Its synthesis, growth mechanism, application in the lithium ion battery, and magnetic and photocatalytic properties. J Phys Chem C 112:4836–4843
Widder KJ, Senyei AE, Scarpelli DG (1978) Magnetic microspheres: a model system for site specific drug delivery in vivo. Proc Soc Exp Biol Med 58:141–146
Garcon G, Garry S, Gosset P, Zerimech F, Martin A, Hannothiaux M, Shirali P (2007) Benzo(a)pyrene-coated onto Fe2O3 particles-induced lung tissue injury: role of free radicals. Cancer Lett 167:7–15
Lawaczeck R, Menzel M, Pietsch HV (2004) Superparamagnetic iron oxide particles: contrast media for magnetic resonance imaging. Appl Organomet Chem 8:506–513
Busch M, Gruyters M, Winter H (2006) Spin polarization and structure of thin iron oxide layers prepared by oxidation of Fe(1 1 0). Surf Sci 600:4166–4169
Walter D (2006) Characterization of synthetic hydrous hematite pigments. Thermochim Acta 445:195–199
Marusak LA, Messier R, White WB (1980) Optical absorption spectrum of hematite α-Fe2O3 near IR to UV. J Phys Chem Solids 41:981–984
Van Krevelen DW, Hoftyzer PJ (1976) Properties of polymers, 3rd edn. Elsevier, Amsterdam
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Rafiee, Z., Golriz, L. Polyimide nanocomposite films containing α-Fe2O3 nanoparticles. J Polym Res 22, 630 (2015). https://doi.org/10.1007/s10965-014-0630-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-014-0630-1