
Abstract
A quantitative description of the molecular networks that sustain morphogenesis is one of the main challenges of developmental biology. In particular, a molecular understanding of the segmentation of the antero-posterior axis in vertebrates has yet to be achieved. This process known as somitogenesis is believed to result from the interactions between a well-studied genetic oscillator and a less established posterior-moving determination wavefront. Here we describe a molecular model for somitogenesis that couples a moving morphogen wavefront with the somitogenetic oscillator. The wavefront is due to a switch between stable states that results from reciprocal negative feedbacks of Retinoic Acid (RA) on the activation of a kinase ErK and of ErK on RA synthesis. We suggest a molecular mechanism by which that switch can be triggered by the somitogenetic clock. The model quantitatively accounts for the shortening of the pre-somitic mesoderm (PSM) in zebrafish in response to the decrease during somitogenesis in the concentration of a morphogen (Fgf8). The generality and robustness of the model allows for its validation (or invalidation) in other model organisms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Among his many contributions to Science, Pierre Hohenberg is famous for his much cited article with Cross in Review of Modern Physics entitled “Pattern formation outside equilibrium” [1]. It is therefore surprising that Pierre did not more forcefully apply his interest in these problems to the quintessential out of equilibrium pattern forming systems found in Biology. Perhaps one of the reasons for this surprising disinterest is to be found in the messiness of biological systems (and the associated proliferation of parameters). As he wrote [1]: “The study of biological patterns involves two basic ingredients: developing a model which incorporates the main mechanisms and properties under consideration, and analyzing the behavior of the model as a function of its parameters. Due to the complexity and richness of the systems studied, the first phase is by far the most challenging and important one for biology, but it is in large measure beyond the scope of our discussion”.
Indeed, in contrast with some expressed opinion, these systems are generally not intelligently designed but arise as a result of tinkering by evolution. While of course constrained by the laws of Physics and Chemistry, Biology often adopt byzantine solutions to engineering problems that are sometimes more akin to Rube Goldberg contraptions than to some nice optimized engineering solution. Somitogenesis provides, we think, a nice illustration of that point.
Somitogenesis is the name given to the segmentation of the antero-posterior (rostro-caudal) axis in vertebrates that precedes the formation of the spinal cord [2]. It begins at the end of gastrulation caudally to the hindbrain with the formation of bilaterally paired segments of mesoderm (the somites) that form periodically along the head-to-tail axis of the growing embryo. In zebrafish this process starts at about 10 h-post-fertilisation (hpf) and ends at 24 hpf, see Fig. 1. The segments appear at regular time intervals from the so-called pre-somitic mesoderm (PSM), the region comprised between the growing tail and the last somite [2]. The time interval between somites’ formation is species specific: about 25 min in zebrafish embryos, 90 min in chicken, 4–5 h in humans. This time interval but not the somite size is strongly temperature dependent. The number of somite pairs generated during somitogenesis differs among various species. There are 31 pairs of somites in zebrafish, 50 in chicken and several hundred in snakes. As precursors of the vertebrae and skeletal muscles, somites are transient structures: their abnormal formation may lead to skeletal and muscular malformations.

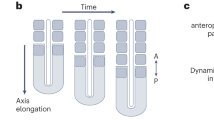
a A zebrafish embryo at 11 somites stage: notice the precursor of the eye, the otic vesicle (ear) and the pre-somitic mesoderm (PSM) from which somites periodically appear. b Kymograph of somitogenesis from 7 to 20 somites (the x-axis is the antero-posterior coordinate). The tail elongates at a constant rate Vtail while the PSM shrinks at a constant rate VPSM resulting in a wavefront of somitogenesis propagating (in the lab frame) at a rate Vfront = Vtail − VPSM
A priori, somitogenesis falls nicely into what Pierre called a type Is instability: the propagation of a stable periodic structure into a uniform one. The period of the stable structure might be set (as in reaction–diffusion patterns) by the interplay between short range excitation and long range inhibition and the propagation of the pattern into the PSM by the gradient of some morphogen controlling the stability of the uniform state. We know of many examples of this type of instability (Rayleigh-Bénard convection, solidification patterns, etc.) and one might have expected an “intelligent designer” to apply similar principles to somitogenesis.
This however is not the case. Instead the following complex mechanism has apparently been selected by evolution. The formation of somites is divided into two main phases. The first phase is the establishment of a pre-pattern in the anterior PSM defined by a stripe of gene expression that sets the place of the somite formed during the next phase. This pre-pattern is a result of the interaction between a genetic oscillator and a posterior-moving wavefront which triggers the cell to differentiate. This Clock and Wavefront model, first advanced by Cooke and Zeeman in 1976 [3], proposes that periodic genetic oscillations (the segmentation clock) couple with a determination front moving posteriorly [2] and stop oscillating. In this model, the size of a somite is determined by the distance travelled by the determination front during one cycle of the segmentation clock [2], see Fig. 2a.

a Clock and Wavefront model: antagonistic gradient of Fgf8 (originating from the posterior PSM, green) and Retinoic Acid (RA, originating from the somites, violet) define a wavefront which interacts with a particular phase of the segmentation clock (in the PSM, red) to generate somites at periodic times and positions. b A simplified version of the somitogenetic clock in zebrafish. Oscillations are due to the time delayed negative feedback of homo or heterodimers of the clock genes Her7 and Her1 (Color figure online)
The second phase of somitogenesis is differentiation into epithelial cells [4] and formation of the morphological boundaries. Cells in the posterior PSM are motile (mesenchimal) and the tissue is thus more fluid. As they move to the anterior PSM they slow down [5] and stop past the determination front. The pattern of gene expression initiated by the interaction between the clock and the wavefront triggers a transition to epithelial cells that form a solid tissue, held together by adhesion molecules. In that phase mechanical tensions in the newly formed tissue result in the formation of a visible furrow between adjacent somites [6, 7], see Fig. 1a.
In support of this model, a segmentation clock coupled to the timing of somite formation has been discovered in many species (see [2, 8]). The implementation of the clock varies across species, presumably a result of the different timescales of somitogenesis. Thus in zebrafish, the clock is formed by the delayed feedback (due to transcription and translation) of some genes (her1, her7) on their own transcription, see Fig. 2b. In amniotes, such as chick and mouse, the clock is implemented by inhibition of Notch signaling by Lfng (lunatic fringe), which is under positive control of NICD (the Notch Intracellular Domain). The overall picture is however more complex, as many proteins are coupled to these oscillations and ensure the stability and synchrony of oscillations between nearby cells (via the Delta/Notch system) [2, 9]. Notice however that the clock frequency is very sensitive to temperature: in zebrafish the time lapse between the formation of adjacent somites can change by a factor two for a change of temperature of only 10 °C. Surprisingly, this extreme sensitivity of the timing of somitogenesis is not reflected in its spatial period: the size of the somite is independent of temperature.
In contrast with the segmentation clock whose details are species dependent, the main actors of the wavefront (e.g. Fibroblast Growth Factors (FGF), Retinoic Acid (RA)) are conserved in vertebrates (from fish to mammals, including snakes and amphibians, see [10]). Many studies have shown that a Fgf8 mRNA gradient forms along the PSM peaking at the posterior part [2, 5, 11, 12]. As cells exit from the PSM, they stop transcribing FGF genes. Thus FGF mRNA progressively decays as cells move towards the anterior of the PSM and an FGF gradient is formed [13]. This FGF mRNA gradient is subsequently translated into a protein gradient and into a kinase activity gradient along the PSM (as Erk, a protein kinase is activated downstream from the FGF receptor) [5, 12], see Fig. 3a.

Schematics of the Fgf signaling network (a) and Retinoic Acid (RA) synthesis pathway (b). a Fgf binds to its receptor (FGFR) and initiates a chain of phosphorylation reactions, which culminates in the phosphorylation of a protein kinase, Erk. This protein translocates to the nucleus and phosphorylates a number of transcription factors which in turn alter the cell state and trigger its differentiation. b Retinoic Acid is synthesized from retinol (Vitamin A), by two oxidation steps, first into retinal and then into RA. RA can be exported in the extracellular medium, where it acts as a morphogen. Alternatively, it can be sequestered in the cytoplasm by CRABP or degraded by CYP26 proteins
Retinoic Acid (RA) was also proposed to be implicated in positioning the determination wavefront [14, 15]. Opposed to the FGF gradient, RA signaling displays an anterior-to-posterior gradient in the anterior-part of the PSM. RA was found in the segmented somites and the anterior-most part of PSM. It is absent in the tail bud and the posterior PSM [16]. RA is synthesized in two sequential oxidation steps from Vitamin A (retinol). The first one, catalyzed by retinal dehydrogenases, converts retinol to retinal, which is then converted into RA by the catalytic action of retinaldehyde dehydrogenases such as RALDH2 [17]. RA can be degraded and inactivated by oxidation to 4-hydroxy-RA which is catalyzed by the cytochrome Cyp26 [18], see Fig. 3b. During somitogenesis, RALDH2 is expressed in the somites and the anterior-most part of PSM while Cyp26 is expressed in the posterior PSM and tail bud [19, 20] and is up-regulated by FGF signaling [21]. An anterior to posterior gradient of RA is thus established in the PSM.
Based on these data and others, Goldbeter, Gonze and Pourquié [22] proposed a model (the G2P model) whereby the mutual inhibition of RA and FGF would result in a bistability of the FGF pathway. The differentiation of the PSM into somites would then be triggered by the clock-induced transition of cells from a high FGF activated state into a low FGF activated one. In their model RA acts to repress the translation of the Fgf8 mRNA gradient into protein (or equivalently induce the degradation of the protein), while Fgf8 activates the degradation of RA via the activation of Cyp26, see Fig. 4a. As a result of this mutual inhibition, the concentration of Fgf8 protein (or equivalently the activity of the protein kinase (Erk) downstream of its receptor) displays a bistable behavior: for a certain range of Fgf8 mRNA concentrations (i.e. in a certain positional window) the kinase activity can be either high or low, see Fig. 2c. Differentiation into somites is then supposed to be triggered (at a certain phase of the segmentation clock) by the inactivation of Erk in that positional window, i.e. by a shift of the Erk activity domain from a rostral position (the limit of the high activity domain) to a more caudal position (the limit of the low activity domain).

a Schematics of the G2P model: RA inhibits translation of the Fgf8 mRNA into protein and Fgf8 induces degradation of RA via Cyp26. b Schematics of the mG2P model: RA inhibits the activity of Erk (via Mkp3, pathway 4) and Erk activates the degradation of RA (via Cyp26, pathway 2). The known positive feedbacks of RA on Fgf8 (pathway 3) and Fgf8 on RaldH (pathway 1) are also included
However the details of the G2P model are contradicted by our own and published data [23]: RA positively controls Fgf8 and similarly Fgf8 positively controls RaldH [16]. To take into account these facts we hereby propose “a model which incorporates the main mechanisms and properties under consideration” [1]. Our model retains the basic feature of the G2P model, namely the bistability of the FGF pathway which it ensures by the negative feedbacks of RA on Erk (mediated by Mkp3 (Dusp6), [24]) and of Erk on RA (mediated by Cyp26; [16, 21], see Fig. 4b. In the following we will describe this modified (mG2P) model and discuss its predictions. A comparison with experimental data will be published elsewhere [27].
2 Results
2.1 An Updated Model for the Somitogenetic Wavefront
The great value of the G2P model is to propose a dynamical molecular model for the interaction network between the main known morphogens implicated in somitogenesis: Fgf8 and Retinoic Acid (RA). This allows for a test of the model by a perturbation of these morphogens and their effectors or targets. For example the model posits a negative feedback of Fgf8 on RA and vice versa, see Fig. 4a. However we and others observed that in contradiction with this assumption, RA correlates positively with the expression of Fgf8 [23, 27]. Similarly it was shown [16, 23] that Fgf8 feeds back positively on the expression of RaldH (the enzyme controlling the synthesis of RA). Hence even though the G2P model provides for a reasonable explanation of somitogenesis, its details are therefore not supported by the experimental data. As noticed by Pierre [1]: “…even if a model accounts reasonably well for the observed phenomena, the molecular picture on which it is founded could be quite incorrect, since the cooperative behavior is often insensitive to the underlying mechanisms”.
Here we propose that the putative bistability window of the Fgf pathway can be implemented at the protein kinase level. Erk, is activated (through phosphorylation, see Fig. 3a, [5]) upon binding of Fgf8 to its receptor (FgfR). Erk is also known to be inactivated (dephosphorylated) by Mkp3 (Dusp6), see Fig. 3a, which is under positive control by RA [15]. Similarly, Erk is implicated in the inhibition of RA [5] via its positive control on Cyp26, the RA degrading enzyme [21]. Hence the bistability of the Fgf pathway is ensured by the mutual inhibition of RA on Erk and vice versa. A dynamical molecular model implementing these known observations (hereafter referred to as the modified G2P (or mG2P) model) is shown in Fig. 4b. Like the G2P model, it displays a window of bistability along antagonistic gradients of RaldH and Fgf8 mRNA. The presence of a bistability window is very robust to changes in the parameters of the model (some of which can be affected by specific drugs) and to external perturbations of RA and Fgf8.
The various models of somitogenesis are usually set in the moving frame of the growing tail, where the concentration of morphogens while space dependent are assumed to be time invariant. As a result as somites appear periodically, the size of the PSM remains constant. This however stands in contradiction with the observations of a varying size of the PSM and an ultimately shrinking PSM. In particular in zebrafish, the PSM is observed to shrink at constant rate. That time variation of the PSM, which can be attributed to time variation of the morphogen gradients, provides a unique test of the validity of the somitogenetic models as their prediction of the variation of the PSM size depends sensitively on the time dependence of the morphogen gradients. To the best of our knowledge that point has been lost and the correlation between the PSM shrinkage and the morphogens time dependence never theoretically investigated nor experimentally studied.
2.2 The mG2P Model
In the moving frame of the tail, the equations describing the molecular network shown in Fig. 4b are as follows. Retinoic acid [RA] is synthesized from retinal by Retinal dehydrogenase (RaldH) at rate vsr and degraded at rate kdr by Cyp26 (at concentration C) and at a basal rate measured by the kinetic constant kd1.
The reported positive feedback of the active (phosphorylated) form of ErK (at concentration EKa) on RaldH (see pathway 1 in Fig. 4b; [16, 23]) is implemented as a Hill equation with half occupation concentration Ekr and a non-cooperative binding (Hill coefficient = 1):
The mRNA of Cyp26 (mC) is under positive feedback of EKa (pathway 2 in Fig. 4b) and is degraded at rate kd2.
where we assumed following [22] a non-linear (n = 2) Hill equation. This non-linearity is essential for bi-stability.
Cyp26 (at concentration C) is synthesized from its mRNA at rate ks3 and degraded at rate kd3.
Fgf8 (at concentration F) is synthesized from its mRNA (mF) at a rate ks4 and degraded at rate kd4
To implement the observed positive feedback of [RA] on mF (pathway 3 in Fig. 4b) we use a Hill equation with non-cooperative binding of [RA] and half occupation R0.
The core of the network is ErK which is activated (phosphorylated) by the Fgf8 pathway and inhibited by [RA] via its action on Mkp3 (pathway 4 in Fig. 4b):
where EK0 is the total ErK concentration, konF is the Fgf8 dependent rate of ErK phosphorylation (activation) and \( k_{spon}^{0} \) and koff[RA]m are respectively the RA independent and dependent rates of ErK dephosphorylation (inactivation). A non-linear Hill coefficient (m = 2) is required for bi-stability.
These equations were simulated (using MATLAB) and at steady state they can reach one or two stable states depending on the initial conditions and the various parameters. Actually at steady-state the above equations simplify enormously. Defining:
Thus at steady state instead of 18 parameters the system depends on only 6 non-dimensional ones (as can be verified by comparing the results of simulations of Eqs. (1–6) with the steady-state predictions Eqs. (8–10)). In the limit \( 1 \ll r; m1 \ll m_{K} \) (negligible pathways 1 and 3), the above model is formally identical to the G2P model (with ErK playing the role of Fgf8). As noticed by Pierre [1]: “the cooperative behavior is often insensitive to the underlying mechanisms”.
If we let some of the parameters in the above model depend on the distance from the tail end, i.e. in presence of gradients, then there might exist along the PSM a window of bistability where two stable solutions are possible with a high or a low ErK activity (variable mK in Eq. (10)). In the comparison with the experimental data we have assumed following [22] that RalDH and the Fgf8 mRNA displayed a monotonous gradient, i.e.:
where \( 0 < \rho \left( u \right), \varphi \left( u \right) < 1 \) and L is a typical scale assumed to be constant during somitogenesis. For simplicity we have set: \( \varphi \left( u \right) = u \) and ρ(u) = 1 − φ(u). We have simulated the mG2P model with exponential gradients in Fgf8 and RaldH, i.e. \( \varphi \left( u \right) = 1 - e^{{ - u/u_{0} }} \) or with ρ(u) =1 (i.e. in presence of a Fgf8 mRNA gradient only) or in presence of a Cyp26 mRNA gradient (\( k_{sc } \to k_{sc } \varphi (x/L)) \), instead of a RaldH gradient, see Fig. 5. In all these situations a bistability window robustly exists (it does not depend on a fine-tuning of the various parameters). A typical set of parameters (similar to the ones mentioned in [22]) is shown in Table 1.

ErK activity at steady-state from a simulation of mG2P model with the parameters shown in the figures: a with a linear gradients of Fgf8 mRNA (parameter γ) only; b together with a linear gradient of Cyp26 (parameter β); c, d or together with a linear gradient of RaldH (parameter α). Notice the existence in all these simulations of a bistability window
The model was run on MATLAB with the parameters shown in Table 1 (and others) and at steady state it clearly displays a bistability window, see Fig. 5a, d.
A plausible description of somitogenesis is thus obtained: as the tail grows, the front propagates (in the lab frame) and at fixed times (i.e. at a set phase of the somitogenetic clock) the undifferentiated posterior PSM (high ErK activity state, blue curve in Fig. 5a–d) is switched in its bistability window into a somite committed state (low ErK activity state, red curve in Fig. 5a–d). Within the present model (set in the moving frame of the tail) this can easily be accounted for by a wave of ErK RA-independent inhibition reaching the bistability window and switching ErK to its low bistability state. That periodic inhibition could possibly be due to Sprouty [25] or some other inhibitor of the Fgf8 pathway, see Fig. 3a, that is coupled to the clock oscillations [26,27,28]. The ErK activity boundary then periodically jumps to its rightmost stability boundary, see Fig. 6, jumps that have indeed been observed [27, 28].

Simulation of the mG2P model with a time periodic modulation of the RA-independent inhibition of Erk (parameter \(\user2{k}_{{\user2{spon}}}^{\user2{0}}\) in Eq. (6)), which results in periodic jumps of the right most boundary of the Erk activity domain
Until now we have assumed, as is usually done, that all the parameters in the mG2P model are constant (time independent). In this case, in the moving frame of the tail, the position of the wavefront is fixed, i.e. the PSM size is time independent. This however is not the case, as can be seen from Fig. 1b: in zebrafish the PSM shrinks at a roughly constant rate. That observation implies that the parameters in the model are not constant, in particular the gradient of Fgf8 mRNA may decay with time, allowing the wavefront to catch up with the growing tail. Simulations of the model with a time dependent gradient of Fgf8 suggest that only an (exponentially) slowing down of this gradient with time can account for the almost constant rate of shrinkage of the PSM. Indeed estimations of the concentration of Fgf8 mRNA (relative to time independent house-keeping genes, such as RPL13 or β-actine) display an exponential decay with time on a typical time-scale of about 8 somitic periods, see Fig. 7. Rescaling the (arbitrary) spatial extent of simulation data by the size of the PSM at 7 somites (t = 0) and the timescales by the measured decay of Fgf8 mRNA yields a good agreement between the model and the observed data, see Fig. 7b, for a large range of values of the simulation parameters, emphasizing the robustness of this model.

a Variation with time of Fgf8 concentration versus somite stage and fit to an exponential decay past 5 somites stage. b PSM shortening from 7 somites stage (dots and error bars on mean; n = 8) and results (continuous line) of a simulation of the mG2P model (with the displayed parameters and assuming an exponential decay of Fgf8 with the timescale measured in (a))
The shrinkage of the PSM offers a convenient way to test models of somitogenesis. By perturbing the actors of the network shown in Fig. 4b (using drugs, an external concentration of RA or genetic methods), monitoring the shrinkage of the PSM and comparing it to simulation results, the models with various gradients can be tested and falsified. Such a comparison is reported elsewhere [27]. It justifies the use of the mG2P model with opposing gradients of RaldH and Fgf8 mRNA (as commonly assumed) in the description of the somitogenetic wavefront.
3 Discussion
We have presented a detailed study of a model of the somitogenetic wavefront which incorporates many known results under a common theoretical description. The proposed molecular model adopts the concept of bi-stability from the original G2P model [22]. It incorporates elements from Moreno and Kintner (the positive feedback of RA on Mkp3 and Fgf8; see also [23]) and Shimozono et al. (the positive feedback of Fgf8 on RaldH) and puts at its core a known kinase of the Fgf pathway, Erk which switching from phosphorylated to dephosphorylated state has been amply documented as a key step in early differentiation into somite fate [5, 10, 28]. In the proposed model the bi-stability is a result of the mutual inhibition between RA and ErK.
While the topology of the network is founded on many different studies, the resulting dynamics has not been thoroughly investigated. Here we describe the predictions of a model of this network, while in a separate publication [27] we present the result of perturbative experiments aimed at testing (falsifying) this model in zebrafish.
Within the mG2P (or G2P) models of somitogenesis with constant (time independent) parameters, the PSM size is constant, in contradiction with observations. To account for the observed almost constant rate of shrinkage of the PSM in zebrafish, the mG2P model requires the Fgf8 mRNA at the tail end to decay exponentially on a timescale larger than the relaxation time (less than a somitic period) of the network describing the wavefront (Fig. 4b). Using qPCR to study experimentally the variation with time of the Fgf8 mRNA we observed that it indeed decayed exponentially with a long timescale (about 8 somitic periods, i.e. ~ 4 h) in agreement with the model. In this adiabatic approximation, the mG2P model can be reduced to a dimensionless form that depends on 6 parameters only (instead of 18 for the full dynamics). Variation of some of these parameters with distance from the tail (e.g. gradients of RaldH and Fgf8 mRNA) results in the possibility of having two co-existing stable states (high and low ErK activity) in a certain range of distances from the tail end that define the position of the wavefront.
A sensible critique of this approach is that with 6 parameters the mG2P model could be fiddled to fit any data. Fortunately, that is not the case as the dynamics of the PSM shrinkage in this model is quite robust (or insensitive) to variation of the parameters. For example, with a linear decrease with time of the Fgf8 mRNA, we could not find parameters for which the PSM shrinks almost linearly with time. The model can thus be used to falsify various hypotheses.
For example a linear gradient of Fgf8 mRNA only can sustain bistability (see Fig. 5a) and could account for the formation of somites in absence of an RA gradient. However the almost constant rate of shrinkage of the PSM observed in WT embryos is incompatible with a linear gradient of Fgf8 mRNA only.
Due to its insensitivity to variation of its parameters, the model can be easily falsified (i.e. it cannot be fiddled to fit any data). For example in chicken and snake the PSM extension varies in a non-monotonous fashion (it increases and then decreases; [10]). Within the present model this result suggests that the Fgf8 mRNA could initially increase and then decrease. By measuring the time dependence of the various actors of the network (in particular the Fgf8 mRNA), and using that data as an input to a simulation of the mG2P model (as we have done here with the observed exponential decay of Fgf8 mRNA) one could compare the simulated PSM dynamics to the observed one and test for the validity of the mG2P model in these other organisms.
Finally, it is interesting to notice that somitogenesis in contrast with physico-chemical pattern forming systems (Rayleigh Bénard, Belousov-Zhabotinsky, etc.) is not characterized by a type Is instability (using Pierre’s nomenclature). Instead the evolved solution appears to imply the interaction between a moving front of antagonistic morphogens (RA and Fgf8) coupled to a segmentation clock located in the growing tail and defining the timing of somite formation. Antagonistic interactions between RA (high in the anterior part of the embryo) and ErK (active in the posterior end) have been reported in an invertebrate chordate where they control posterior patterning [29]. It is tempting to speculate that in vertebrates, segmentation evolved via the inhibition of this primitive network by an independent pulsating one (the segmentation clock) which details may have evolved differently in different organisms.
References
Cross, M.C., Hohenberg, P.C.: Pattern formation outside of equilibrium. Rev. Mod. Phys. 65, 851–1112 (1993)
Dequéant, M.-L., Pourquié, O.: Segmental patterning of the vertebrate embryonic axis. Nat. Rev. Genet. 9, 370–382 (2008). https://doi.org/10.1038/nrg2320
Cooke, J., Zeeman, E.C.: A clock and wavefront model for control of the number of repeated structures during animal morphogenesis. J. Theor. Biol. 58, 455–476 (1976). https://doi.org/10.1016/S0022-5193(76)80131-2
Kulesa, P.M., Fraser, S.E.: Cell dynamics during somite boundary formation revealed by time-lapse analysis. Science 298, 991–995 (2002). https://doi.org/10.1126/science.1075544
Delfini, M.-C., Dubrulle, J., Malapert, P., Chal, J., Pourquié, O.: Control of the segmentation process by graded MAPK/ERK activation in the chick embryo. Proc. Natl. Acad. Sci. U.S.A. 102, 11343–11348 (2005). https://doi.org/10.1073/pnas.0502933102
Durbin, L., Brennan, C., Shiomi, K., Cooke, J., Barrios, A., Shanmugalingam, S., Guthrie, B., Lindberg, R., Holder, N.: Eph signaling is required for segmentation and differentiation of the somites. Genes Dev. 12, 3096–3109 (1998). https://doi.org/10.1101/gad.12.19.3096
Pourquié, O.: Vertebrate somitogenesis. Annu. Rev. Cell Dev. Biol. 17, 311–350 (2001)
Schröter, C., Herrgen, L., Cardona, A., Brouhard, G.J., Feldman, B., Oates, A.C.: Dynamics of zebrafish somitogenesis. Dev. Dyn. 237, 545–553 (2008)
Pourquié, O.: Vertebrate segmentation: from cyclic gene networks to scoliosis. Cell 145, 650–663 (2011). https://doi.org/10.1016/j.cell.2011.05.011
Gomez, C., Ozbudak, E.M., Wunderlich, J., Baumann, D., Lewis, J., Pourquié, O.: Control of segment number in vertebrate embryos. Nature 454, 335–339 (2008). https://doi.org/10.1038/nature07020
Dubrulle, J., McGrew, M.J., Pourquié, O.: FGF signaling controls somite boundary position and regulates segmentation clock control of spatiotemporal Hox gene activation. Cell 106, 219–232 (2001)
Sawada, A., Shinya, M., Jiang, Y.J., Kawakami, A., Kuroiwa, A., Takeda, H.: Fgf/MAPK signalling is a crucial positional cue in somite boundary formation. Development. 128, 4873–4880 (2001)
Dubrulle, J., Pourquié, O.: fgf8 mRNA decay establishes a gradient that couples axial elongation to patterning in the vertebrate embryo. Nature 427, 419–422 (2004). https://doi.org/10.1038/nature02216
Diez del Corral, R., Olivera-Martinez, I., Goriely, A., Gale, E., Maden, M., Storey, K.: Opposing FGF and retinoid pathways control ventral neural pattern, neuronal differentiation, and segmentation during body axis extension. Neuron 40, 65–79 (2003)
Moreno, T.A., Kintner, C.: Regulation of segmental patterning by retinoic acid signaling during xenopus somitogenesis. Dev. Cell 6, 205–218 (2004). https://doi.org/10.1016/S1534-5807(04)00026-7
Shimozono, S., Iimura, T., Kitaguchi, T., Higashijima, S.-I., Miyawaki, A.: Visualization of an endogenous retinoic acid gradient across embryonic development. Nature 496, 363–366 (2013). https://doi.org/10.1038/nature12037
Niederreither, K., Vermot, J., Le Roux, I., Schuhbaur, B., Chambon, P., Dollé, P.: The regional pattern of retinoic acid synthesis by RALDH2 is essential for the development of posterior pharyngeal arches and the enteric nervous system. Development. 130, 2525–2534 (2003)
Kam, R.K.T., Deng, Y., Chen, Y., Zhao, H.: Retinoic acid synthesis and functions in early embryonic development. Cell Biosci. 2, 11 (2012). https://doi.org/10.1186/2045-3701-2-11
Blentic, A., Gale, E., Maden, M.: Retinoic acid signalling centres in the avian embryo identified by sites of expression of synthesising and catabolising enzymes. Dev. Dyn. 227, 114–127 (2003). https://doi.org/10.1002/dvdy.10292
Sakai, Y., Meno, C., Fujii, H., Nishino, J., Shiratori, H., Saijoh, Y., Rossant, J., Hamada, H.: The retinoic acid-inactivating enzyme CYP26 is essential for establishing an uneven distribution of retinoic acid along the anterio-posterior axis within the mouse embryo. Genes Dev. 15, 213–225 (2001)
Pownall, M.E., Isaacs, H.V.: FGF signalling in vertebrate development. Morgan & Claypool Life Sciences, San Rafael (CA) (2010)
Goldbeter, A., Gonze, D., Pourquié, O.: Sharp developmental thresholds defined through bistability by antagonistic gradients of retinoic acid and FGF signaling. Dev. Dyn. 236, 1495–1508 (2007). https://doi.org/10.1002/dvdy.21193
Hamade, A., Deries, M., Begemann, G., Bally-Cuif, L., Genêt, C., Sabatier, F., Bonnieu, A., Cousin, X.: Retinoic acid activates myogenesis in vivo through Fgf8 signalling. Dev. Biol. 289, 127–140 (2006). https://doi.org/10.1016/j.ydbio.2005.10.019
Moreno, T.A., Jappelli, R., Belmonte, J.C.I., Kintner, C.: Retinoic acid regulation of the Mesp-ripply feedback loop during vertebrate segmental patterning. Dev. Biol. 315, 317–330 (2008). https://doi.org/10.1016/j.ydbio.2007.12.038
Hayashi, S., Shimoda, T., Nakajima, M., Tsukada, Y., Sakumura, Y., Dale, J.K., Maroto, M., Kohno, K., Matsui, T., Bessho, Y.: Sprouty4, an FGF inhibitor, displays cyclic gene expression under the control of the notch segmentation clock in the mouse PSM. PLoS ONE 4, e5603 (2009). https://doi.org/10.1371/journal.pone.0005603
Sari, D.W.K., Akiyama, R., Naoki, H., Ishijima, H., Bessho, Y., Matsui, T.: Time-lapse observation of stepwise regression of Erk activity in zebrafish presomitic mesoderm. Sci. Rep. 8, 4335 (2018). https://doi.org/10.1038/s41598-018-22619-9
Zhang, W., Ducos, B., Delagrange, M., Vriz, S., Bensimon, D.: Quantitative study of the somitogenetic wavefront in zebrafish. Development (2018). https://doi.org/10.1101/419705
Akiyama, R., Masuda, M., Tsuge, S., Bessho, Y., Matsui, T.: An anterior limit of FGF/Erk signal activity marks the earliest future somite boundary in zebrafish. Development 141, 1104–1109 (2014). https://doi.org/10.1242/dev.098905
Pasini, A., Manenti, R., Rothbächer, U., Lemaire, P.: Antagonizing retinoic acid and FGF/MAPK pathways control posterior body patterning in the invertebrate chordate ciona intestinalis. PLoS ONE 7, e46193 (2012). https://doi.org/10.1371/journal.pone.0046193
Acknowledgements
We thank A.Goldbeter and V.Hakim for constructive criticism of our model. This work was partially supported by Grants ANR-10-LABX-54 MEMO LIFE and ANR-11-IDEX-0001-02 PSL* Research University and PSL Grants SuperLINE and MicroGUT.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations
Rights and permissions
About this article

Cite this article
Zhang, W., Mayr, V., Ducos, B. et al. A Model of Somitogenesis. J Stat Phys 175, 729–742 (2019). https://doi.org/10.1007/s10955-019-02265-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10955-019-02265-9