
Abstract
Optimized hydrogen-bond descriptors are provided for 21 primary and secondary alcohols, 16 phenols, 10 anilines and 4 amides derived from experimental chromatographic and liquid partition data analyzed using the solvation parameter model with the Solver method. Solver is an optimization package in Microsoft Excel that simultaneously adjusts the descriptor values to minimize the sum of the standard deviation of the residuals for the dependent experimental variable for a set of model equations using a Simplex algorithm. For the full data set the optimized hydrogen-bond acid descriptors are approximately 1.27 times larger than Abraham’s descriptors and the hydrogen-bond base descriptors roughly equivalent. For an isolated aliphatic primary hydroxyl group the hydrogen-bond acidity was estimated as A = 0.340 and hydrogen-bond basicity B = 0.539 and compared with a secondary hydroxyl group is a stronger hydrogen-bond acid and similar hydrogen-bond base, A = 0.26 and B = 0.54. For alcohols of the type C6H5(CH2)mOH the A and B descriptors are linearly related to m for at least m = 1 to 3. In unsubstituted phenols the hydroxyl group is more hydrogen-bond acidic, A = 0.740, and less hydrogen-bond basic, B = 0.158, than a primary aliphatic hydroxyl group. Ortho substituents reduce the hydrogen-bond acidity of a phenolic hydroxyl group in the order –CH3 < –Cl < < –NO2 with 2-nitrophenol almost non-hydrogen-bond acidic, A = 0.062, due to favorable intramolecular hydrogen-bonding. For para-substituted phenols hydrogen-bond acidity increases in the order –CH3 < –Br < –C6H5 < –CN ≈ –Cl < < –NO2 with a range for A from 0.673 to 0.975 and for hydrogen-bond basicity –Cl ≈ –Br < –NO2 < –CH3 < –CN < –C6H5 with a range for B from 0.775 to 1.267. The ranking order is not simply explained by consideration of inductive or mesomeric effects acting individually. The –NH2 group in aniline is a weaker hydrogen-bond acid, A = 0.238, and stronger hydrogen-bond base than phenol. A single methyl group in any ring position for anilines simultaneously increases the hydrogen-bond basicity and reduces the hydrogen-bond acidity of the –NH2 group. A single nitro group in any ring position has the opposite effect on the hydrogen-bonding properties of the –NH2 group. Primary amides are relatively strong hydrogen-bond acids and bases compared with primary alcohols and anilines and occupy the same descriptor space as phenols. An NMR correlation model is developed to estimate the optimized A descriptor using chemical shift differences for acidic protons in DMSO and CDCl3. The principle of additivity is not a good model for estimating the A and B descriptors for compounds with more than one hydrogen-bond acid/base site, although it provides acceptable results in some cases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
A major contribution to the understanding of the solvation properties of neutral compounds was made by Abraham et al. who systematically derived a model to describe solute–solvent intermolecular interactions based on free energy related solution properties [1,2,3]. Initially, separate scales for hydrogen-bond acidity (\(\alpha_{2}^{{\text{H}}}\)), or hydrogen-bond donor scale and hydrogen-bond basicity (\(\beta_{2}^{{\text{H}}}\)), or hydrogen-bond acceptor scale, were set up based on the measurement of 1:1 equilibrium complexation constants for monofunctional compounds in inert solvents. These equilibrium constants for multiple acids or bases against a common acid or base formed a series of linear plots intersecting at a “magic point” that provided a natural origin for each scale. These were the first free energy related scales of hydrogen bonding with a defined zero and represented an important advance in studies of quantitative hydrogen bonding. These scales, however, fail to account for the properties of compounds with several hydrogen bonding sites in which each site is potentially simultaneously involved in hydrogen-bond acid–base interactions with surrounding solvent molecules as well as intramolecular interactions between sites. New scales of effective or overall hydrogen-bonding were required to describe the properties of a single molecule surrounded by solvent molecules relevant to modeling general solvation properties. Furthermore, several other solute–solvent interactions as well as hydrogen bonding need to be considered, and so various solvation parameters had to be devised. After several preliminary trial-and-error approaches, a model with a consistent set of parameters applicable to the solvation of neutral compounds was devised and is today identified as the solvation parameter model [4,5,6]. The general model using the modern nomenclature is given below for transfer from an ideal gas phase to a solvent
and for transfer between immiscible solvents
The dependent variable, SP, is a free energy related property for multiple solutes in a single solvent, such as retention factors in gas or liquid chromatography or partition constants in biphasic liquid–liquid partition systems. The capital letters are referred to as Descriptors defined in Table 1. The descriptors describe the contribution of the solute to the experimental measurements. The lower-case letters in italics are referred to as System Constants and describe the complementary solvent properties to the descriptors. Initially, Abraham et al. used a stepwise procedure employing gas chromatographic retention data on non-polar stationary phases [7], polar compounds lacking hydrogen-bond acid functional groups on polar stationary phases [8], and polar compounds with hydrogen-bond acid functional groups on polar stationary phases [8] to obtain approximate values for the L, S and A descriptors. None of the common gas chromatographic stationary phases are significant hydrogen-bond acids (b = 0) [9]. To estimate the hydrogen-bond basicity descriptor aqueous liquid–liquid partition constants were utilized [10]. The procedure employed the setup of several equations of the type indicated by Eq. 2 with \(\alpha_{2}^{{\text{H}}}\) and \(\beta_{2}^{{\text{H}}}\) employed as trial values for A and B to estimate initial system constants. New estimates of the hydrogen-bonding descriptors were then obtained by back-calculation, often significantly different from the trial values. These new descriptor estimates were then used to re-calculate the system constants for the models. This process was repeated until a self-consistent set of equations and descriptor values were obtained.
Several databases of experimental descriptors for use with the solvation parameter model are available as subscription software [11], as a free internet resource [12], or from a single laboratory with prescriptive quality control procedures [13]. There are inconsistencies in descriptor values compiled from literature sources often with no indication for which value represents the best estimate of the true value. This arises from the use of different sources and quality of experimental data to calculate the descriptors, and in some cases, even the inclusion of estimated values for part of the experimental data. Descriptor quality is a significant issue and the dispersion of descriptor values effects model precision and the estimate of solvent properties. Ideally, when different compounds are used to calibrate systems for descriptor measurements the descriptors for the calibration compounds should be of comparable quality. The Wayne State University (WSU) experimental descriptor database represents an attempt to provide reference compounds for calibration purposes by employing defined protocols for all experimental measurements with appropriate statistical controls [13]. Collections of varied compounds of uniform descriptor quality have been recommended for calibrating open-tubular columns for gas chromatography [14] and packed columns for reversed-phase liquid chromatography [15, 16] for a wide range of experimental conditions to begin the process of standardizing calibration procedures for system constants and descriptor measurements [17]. Using standardized conditions, the goal of this work is to obtain a more self-consistent and reliable set of compound descriptors suitable for calibrating experimental systems for estimating descriptor values for additional compounds and to explore the relationships between compound descriptors and molecular structure for simple compounds in the hope of minimizing the ambiguity in the interpretation. A secondary goal was to outline the protocol using the Solver method to enable its use by scientists new to this technique who might want to contribute compounds of specific interest to their studies to descriptor databases.
2 Methodology
Equations 1 and 2 for calibrated chromatographic and liquid–liquid partition systems can be used to estimate descriptors for other compounds by solving a set of simultaneous equations with the descriptors as variables and experimental retention factors and partition constants for the compound in all systems as the dependent variable [5, 6, 18, 19]. The Solver algorithm in Microsoft Excel provides a convenient tool for these calculations [20]. Solver is an optimization package that adjusts selected changing cells (the descriptors) to minimize the value in a target cell (the sum of the standard deviation of the observed—predicted value for the dependent variable for all equations). Any calibrated model can be used for descriptor calculations; however, it is beneficial if the chosen systems have one or more relatively large system constants and a small standard error of the estimate for the model to minimize errors in fitting the descriptor values. The V descriptor is always available by calculation and is entered in Solver as a fixed value constraint. The E descriptor for liquids with a refractive index value is also available by calculation and is entered as a fixed value constraint. For solids, E is fitted in the same way as the other descriptors. For the A and B hydrogen-bonding descriptors limiting constrains are applied based on chemical principles (A and B must be ≥ 0). It is important that the range for the dependent variable is roughly scaled to a similar magnitude when using the Solver method. For chromatographic measurements this is rarely an issue as the scale for retention factor measurements is restricted to an acceptable separation time. Liquid–liquid partition constants and other imported data may lie significantly outside this range. In this case the model system constants, c term, and dependent variable are divided by an arbitrary integer to obtain the same approximate range as the other values for the dependent variable.
To solve the system of equations for all descriptors the minimum number of equations (models) required is equal to the number of descriptors that are free to vary. The use of such small datasets is however not advisable and typically larger (over defined) datasets are used for better statistical management and identification of erroneous experimental measurements. Also, the Solver method allows further systems to be added later to improve initial descriptor estimates.
Since the models used to determine the descriptors are themselves estimates, Solver is used to optimize both the system constants and descriptors simultaneously [5, 6, 15, 19]. This process is illustrated in Fig. 1. Initial estimates of the solute descriptors were taken from [18] or [12] and used to estimate the system constants for all models by multiple linear regression analysis for each model in turn. These models were then assembled in the Solver file used to optimize the descriptors for each compound. The optimization then continues in a cyclic fashion until the difference between the last estimate of the descriptors and the previous estimate are statistically insignificant at the 95% confidence level. This process is also applied when additional data in the form of further models is added to the Solver file to update the best estimate of the descriptors. The optimization process can also be applied when literature sources for chromatographic models are used as the initial estimates for the system constants.

General scheme for optimizing descriptors by the Solver method
The experimental data for calculation of the hydrogen-bonding descriptors was assembled from the retention factor or partition constant databases for gas chromatography on open-tubular capillary columns [21, 22], ionic liquid stationary phases [23,24,25,26], and hydrogen-bond acid stationary phases [9, 27]; retention factor databases for reversed-phase liquid chromatography with various mobile phase compositions and organic modifiers [28, 29]; partition constant databases for totally organic biphasic systems [30,31,32]; and partition constant databases for octanol–water [33], chloroform–water [34], 1,2-dichloroethane–water [35], chloroform–methanol–water (Folch partition) [36], cyclohexane–water [37], and hexadecane–water [37]. Initial estimates for the system constants were taken from the same databases for the chromatographic systems and are largely unchanged through the optimization process. The final system constants for the liquid–liquid partition constants are summarized in Table 2.
3 Results and Discussion
Abraham’s hydrogen-bonding descriptors are derived from experimental measurements and subject to errors associated with the quality and quantity of the experimental data [5, 6, 13, 18]. By using a saturated statistical experimental design and strict experimental protocols we hope to replace the original hydrogen-bonding descriptors with a database of more accurate descriptors [13]. The hydrogen-bonding descriptors are given for infinite dilution conditions, that is a single solute surrounded by solvent molecules. This is a reasonable model for gas and liquid chromatographic retention factor measurements, but liquid–liquid partition measurements are always made at finite concentration to allow detection of the solute in both phases. For solutes with partition constants (log10 K) > ± 3 this may be a problem since the solute concentration has to be adjusted to allow detection in the phase containing the lowest solute concentration. This was an issue for some of the biphasic aqueous systems described in Sect. 2. Since these partition constants were not determined in our laboratory, and we have inadequate information to evaluate the quality control procedure used in their measurement, we arbitrarily removed any data for partition constants for the aqueous biphasic systems in which the difference in the model predicted and experimental values exceeded two standard deviations in the Solver method.
Several methods have been proposed to estimate Abraham’s hydrogen-bonding descriptors based on the principle of additivity of atom or fragment constants [2, 38,39,40,41] or use quantum chemical molecular descriptors [42,43,44,45,46]. These models regress a set of parameters related to molecular structure against the experimental hydrogen-bonding descriptors with a view to reproducing the experimental values with minimum error, and as such, depend upon having accurate experimental values for the hydrogen-bond descriptors. They also highlight the difficulty of accommodating electronic effects (inductive and mesomeric), steric, and intramolecular hydrogen-bonding in models employing the principle of additivity. It is these issues together with measurement uncertainty in the hydrogen-bond descriptors that limits models based on group contribution approaches to estimation methods. Yet these methods can still prove useful in delineating the contribution of molecular structure to the hydrogen-bonding properties of functional groups.
3.1 Aliphatic and Aromatic Alcohols
The descriptors for several primary aliphatic and aromatic alcohols either belonging to a homologous series or with a single structural variation with the potential to modify the hydrogen-bonding properties of the hydroxyl group through steric or inductive effects are summarized in Table 3. n-Alkanes have no dipolarity/polarizability or hydrogen-bonding capability (A = B = S = 0). For the aliphatic primary alcohols, it is reasonable to assume that the A, B and S descriptors result solely from the contribution of the hydroxyl group in the absence of steric effects and solute–solute interactions [3, 10]. Also, it is reasonable to consider the hydroxyl hydrogen as the only hydrogen-bond acid site and the lone pair electrons on the hydroxyl group as the only hydrogen-bond base sites in this case. For the six aliphatic n-alcohols in Table 3, the average descriptor values (standard deviation in brackets) are S = 0.433 (0.012), A = 0.340 (0.011) and B = 0.539 (0.006). If these values are taken as the best estimate of the true value for the descriptors, then the experimental uncertainty can be estimated by the average absolute error (AAE) as 0.009 for S, 0.008 for A, and 0.006 for B. These values are considerably smaller than the AAE values cited by Abraham and workers at around 0.01 to 0.05 [2, 3, 8, 10]. For an aliphatic primary hydroxyl group Abraham assigned the values S = 0.42, A = 0.37 and B = 0.48, in poor agreement with the optimized descriptors determined here, particularly for the B descriptor. A comparison of 2-methylbutan-1-ol and 3-methylbutan-1-ol with pentan-1-ol, Table 3, indicates no significant difference in hydrogen-bond acidity associated with chain branching but small differences in dipolarity/polarizability and hydrogen-bond basicity possibly associated with minor changes in the cavity shape. For 2-methylpropan-1-ol differences in the dipolarity/polariability are enhanced but the hydrogen-bonding descriptors are effectively unchanged.
1,12-Dodecanediol was evaluated to establish whether the contribution of a hydroxyl group to solute–solvent interactions could be considered additive in aliphatic compounds where the hydroxyl groups are incapable of intramolecular hydrogen bonding. For two insolated primary hydroxyl groups the estimated values are S = 0.806, A = 0.704 and B = 1.036 assuming an additive model compared with the experimental values S = 0.894, A = 0.690 and B = 1.433. The A descriptor is approximately additive, in relative terms the difference between the estimated and experimental value is about 2%, but larger differences are observed for the S and B descriptors, at about 10% and 28%, respectively. It might be reasonable to use the principle of additivity to estimate the A descriptor for alkanediols with isolated hydroxyl groups, but this is not the case for the S and B descriptors.
For the three aromatic primary alcohols of the type C6H5(CH2)mOH the principle of additivity of the two separate hydrogen-bonding sites C6H5– and –OH does not hold for the S, A or B descriptors. Taking the descriptors for benzene as a suitable model for the C6H5– fragment we have S = 0.512, A = 0, and B = 0.140 [13]. Since benzene is a non-hydrogen-bond acid, then in the absence of electronic interactions between the phenyl ring and the hydroxyl group, the estimated value for A would be the same as for a primary aliphatic hydroxyl group, or A = 0.34. Instead, there is a clear linear trend in the A descriptor in which the hydrogen-bond acidity of the aromatic primary alcohols decreases with increasing m without reaching the predicted value for a primary aliphatic alcohol, at least for m = 1 to 3
Here and elsewhere in this report r2 is the coefficient of determination, SE the standard error of the estimate, n the number of values for the dependent variable, and F is the Fisher statistic. The standard deviation of the regression coefficients is not shown for Eq. 3 as these are vanishingly small. These results suggest that electronic interactions resulting from the introduction of a phenyl group in compounds of the type C6H5(CH2)mOH are not fully suppressed for m up to 3, or alternatively, there may be differences in the cavity shape that effects the solvation shell when a phenyl group is substituted for an alkyl group.
The hydrogen-bond base descriptor exhibits a similar trend with an opposite sign to the A descriptor for compounds of the type C6H5(CH2)mOH
The replacement of a methyl group by a phenyl ring simultaneously increases the hydrogen-bond basicity and decreases the hydrogen-bond acidity of the aromatic primary alcohols as a linear function of the number of methylene groups, at least for the alcohols with m = 1 to 3. Interestingly, the S descriptor is essentially constant for m = 1 to 3 at 0.855 but significantly smaller than the value estimated for summation of a benzene ring and a hydroxyl group at 0.945.
Cinnamyl alcohol (3-phenylprop-2-en-1-ol) differs from 3-phenylpropan-1-ol in having a double bond at the C-2 position in the alkyl chain capable of extending possible electronic interaction between the phenyl ring and hydroxyl groups. Cinnamyl alcohol is a stronger hydrogen-bond acid and weaker hydrogen-bond base than 3-phenylpropan-1-ol, and a significantly stronger hydrogen-bond acid than an aliphatic primary alcohol, see Table 3. Although the olefinic group itself is a non-hydrogen-bond acid and weak hydrogen-bond base, its ability to promote electronic communication between the phenyl ring and hydroxyl group has a significant effect on the hydrogen-bonding properties of cinnamyl alcohol. Taking styrene as a model for the phenylvinyl fragment (S = 0.660, A = 0, and B = 0.173 [13]) there is poor agreement for the estimated hydrogen-bonding properties of cinnamyl alcohol assuming an additive model, but reasonable agreement for the dipolarity/polarizability descriptor (S = 1.003 compared with the estimated value 1.093). For 2-(1-naphthyl)ethanol a similar analysis indicates that that the A and S descriptors are not additive using naphthalene as a model for the naphthyl group (S = 0.919, A = 0 and B = 0.190 [13]). While the principle of additivity proves reasonable for the hydrogen-bond basicity descriptor (B = 0.751 compared with the estimated value of 0.729) the A and S descriptors show only poor agreement.
The effect of the electron density on the phenyl ring on the hydrogen-bond acidity of the hydroxyl group of benzyl alcohol can be evaluated by introducing an electron withdrawing (nitro group) or electron donating (methoxy group) at the para position on the benzene ring. The methoxy group in 4-methoxybenzyl alcohol (A = 0.538) is more effective than the nitro group in 4-nitrobenzyl alcohol (A = 0.497) at enhancing the hydrogen-bond acidity of the hydroxyl group of benzyl alcohol (A = 0.429). However, given that both substituents increase the hydrogen-bond acidity of the hydroxyl group the situation is more complicated than can be explained from straightforward assumptions based on the relative π-electron density on the phenyl ring. Since the nitro and methoxy groups are themselves both dipolar/polarizable and hydrogen-bond basic there is no simple model to explain the changes in the S and B descriptors without making speculative assumptions.
Octan-2-ol, cyclohexanol and dihydrocholesterol were taken as representative examples of a secondary alcohol with a hydrocarbon backbone. The descriptor wells for the hydrogen-bond acid descriptor are shown in Fig. 2. The construction of the descriptor wells is described in the figure legend with further information in [18, 20]. The bottom of the descriptor wells is relatively flat for the three secondary alcohols with only a small variation of the standard deviation over the range A ≈ 0.25 to 0.30. To obtain the best estimate of the true value for the A descriptor the fixed constraints in Solver were used to set target values for the A descriptor with the other descriptors allowed to vary unless their true value was available by calculation (V for all secondary alcohols and E for secondary alcohols with an experimental refractive index value, see Table 1). The results are summarized in Table 4. Considering the minimum value of the descriptor wells for the three secondary alcohols the preferred value for A is 0.26. There is some variation in the B descriptor for the selected A value and the average for the three alcohols, 0.54 (± 0.03), is taken as the best estimate of the true value. Thus, an aliphatic secondary hydroxyl group is less hydrogen-bond acidic than a primary aliphatic hydroxyl group (A = 0.34 compared with 0.26), about as hydrogen-bond basic (B = 0.539 compared with 0.54) and dipolar/polarizable (S = 0.427 compared 0.433 for octan-2-ol). Both carbon skeletons for cyclohexane and dihydrocholesterol are slightly dipolar/polarizable, S = 0.10 and 0.414, respectively, and the S descriptor cannot be estimated by assuming the principle of additivity of fragment groups with cyclohexane and cholestane as models for the carbon skeleton, unlike 2-octanol for which S = 0 for n-octane.

Descriptor wells for the hydrogen-bond acid descriptor of some secondary alcohols. Identification: 1 = cyclohexanol, 2 = octan-2-ol, and 3 = dihydrocholesterol. The wells are constructed by applying fixed constraints in Solver for those descriptors available by calculation, Table 1, with the other descriptors free to vary subject to chemical constraints (S, A and B ≥ 0) for a series of hypothetical values for A selected to span the minimum value of A observed for the descriptor well
Triisopropanolamine contains three secondary aliphatic hydroxyl groups in close proximity that might support intramolecular hydrogen-bonding resulting in a diminished capability for solute–solvent intermolecular interactions. The optimized descriptors for triisopropanolamine are:
For three independent secondary hydroxyl groups the estimated value for A = 0.78 and for B = 1.62 assuming the principle of additivity and ignoring contributions from the nitrogen atom core to the effective hydrogen-bond basicity for convenience. The experimental hydrogen-bond acidity is about 45% of the estimated value inferring that intramolecular hydrogen-bonding likely contributes to the solvation properties of triisopropanolamine. The hydrogen-bond basicity is about 94% of the estimated value and would suggest that intramolecular hydrogen-bonding is far less effective at suppressing solute hydrogen-bond base interactions with surrounding solvent.
A systematic deviation for the experimental and predicted octanol–water partition constants was observed for the aliphatic alcohols without a phenyl group in the alkyl chain, Table 5. The model for the octanol–water partition constant is given in Table 2. The partition constants (log10 KOW) are about 15% larger than those predicted using the descriptors in Tables 3 and 4. No systematic deviations were observed for these compounds in other biphasic liquid–liquid partition systems. This suggests a phase-specific interaction with n-octanol resulting in an increase in solubility of the aliphatic alcohols in the octanol-phase possibly because of more favorable hydrogen-bonding interactions between alkyl alcohols and n-octanol.
3.2 Phenols
Simple phenols have an aromatic core with functional groups in well-defined positions, which can interact through space if located on adjacent carbon atoms and electronically over longer distances through inductive and mesomeric effects. As model systems for hydrogen-bonding interactions they have been studied numerous times using spectroscopic, calorimetric, partition and theoretical calculation methods [2, 47,48,49,50,51,52,53]. The aromatic core for the phenols considered here, can be modeled by benzene, naphthalene, and biphenyl, all of which are non-hydrogen-bond acids and weak hydrogen-bond bases, B = 0.140, 0.190 and 0.278, respectively [13]. As shown in Table 6, the hydrogen-bond acidity of phenol, 1-naphthol and 2-naphthol are essentially identical with A = 0.740, and not much different for 4-phenylphenol, A = 0.799. Thus, A = 0.740 is a reasonable estimate for the hydrogen-bond acidity of a single phenolic hydroxyl group in the absence of electronic and steric effects and is a considerably stronger hydrogen-bond acid than a typical primary aliphatic alcohol, A = 0.340. The hydrogen-bond basicity of the phenolic group can be estimated as 0.158 (0.017) assuming additivity of the phenol group and aromatic core for the four phenols indicated above and is a significantly weaker hydrogen-bond base than a primary aliphatic alcohol, B = 0.539. In a similar manner, the dipolarity/polarizability of the phenolic hydroxyl group can be estimated as 0.256 (0.032), which is smaller than for a primary aliphatic alcohol, S = 0.433.
The methyl group is a weak electron donor and when located next to a phenolic hydroxyl group could interfere in hydrogen-bond intermolecular interactions because of intramolecular hydrogen-bond formation or due to steric factors. The descriptors for 2-, 3-, and 4-methylphenol and 2,6-dimethyphenol are given in Table 6. An ortho methyl group reduces the hydrogen-bond acidity of phenol by about 0.1 units and two ortho methyl groups by about 0.3 units. For monomethyl phenols there is not much difference in hydrogen-bond acidity for phenols with an ortho or para methyl group, about 0.04 units, which could be due to electronic factors as much as steric considerations or formation of an intramolecular hydrogen-bond. This is also possibly true for 2,6-dimethylphenol, except that the larger change in hydrogen-bond acidity is difficult to explain solely by electronic effects. The hydrogen-bond basicity of the monomethylphenols shows little change with structure and is only slightly greater than phenol itself. The dipolarity/polarizability for the methyl-substituted phenols is like phenol itself with an average of S = 0.752 (0.018, n = 5) without any obvious structural dependence. The S, A and B descriptors for 1,3-dihydroxybenzene are approximately additive S = 1.024 (predict 0.995), A = 1.376 (predict 1.480) and B = 0.485 (predict 0.456) suggesting that the phenolic groups are isolated and electronic effects are small.
Phenols containing ortho substituents with a greater capability than the methyl group to participate in intramolecular hydrogen-bond formation include 2-chlorophenol and 2-nitrophenol [48,49,50,51]. The formation of an intramolecular hydrogen-bond is expected to diminish the capability of the phenol hydroxyl group to participate in intermolecular hydrogen bonding. For the ortho substituted phenols the hydrogen-bond acidity follows the order –CH3 < –Cl < < –NO2. 2-Nitrophenol is almost non-hydrogen-bond acidic, A = 0.062, and when compared with 3- and 4-nitrophenol with A > 1.0, suggests that the proximity of the functional groups favors intramolecular hydrogen-bonding. 2-Chlorophenol is intermediate with A = 0.530 and not as extensively involved in intramolecular hydrogen bonding but still considerably less hydrogen-bond acidic than phenol itself, A = 0.740. Spectroscopic evidence indicates that 2-chlorophenol exists as a mixture of cis- and trans-conformers in which only the cis-form can participate in an intramolecular hydrogen-bond [51]. Steric and electronic effects may also be important and for 2-chlorophenol but it cannot be implied that the observed reduction in hydrogen-bond acidity is due solely to intramolecular hydrogen bonding. The intrinsic hydrogen-bond basicity of the –CH3, –Cl, and –NO2 functional groups is unknown but the range for the B descriptor (0.34 to 0.38) for the three ortho-substituted phenols does not indicate significant variation when compared with the A descriptor. 2-Nitrophenol is considerably more hydrogen-bond basic than either the 3- or 4-nitrophenols probably largely because of intramolecular hydrogen bond formation. There are large changes for the S descriptor mirroring expectations based on dipole moments for the ortho-substituted phenols [48]. All three nitrophenols are dipolar/polarizable with 3-nitrophenol the most dipolar/polarizable, S = 1.246, and 4-nitrophenol the least, S = 0.975. These results are likely due to electronic effects and not obviously influenced by intramolecular hydrogen-bonding in the case of 2-nitrophenol.
The contribution of electronic effects to the hydrogen-bond acidity of the phenol hydroxyl group can be assessed from the descriptors for the para-substituted phenols. For the A descriptor the order is –CH3 < –Br < –C6H5 < –CN ≈ –Cl < < –NO2 with a range 0.673 to 0.975. Since none of the substituents are hydrogen-bond acids the order must reflect electronic effects but is not simply explained by correlation with the electron donor/acceptor properties of the substituent groups. It is likely that inductive and mesomeric effects operate simultaneously to enhance some and reduce other functional group interactions with the phenolic hydroxyl group. 4-Methylphenol and 4-bromophenol are weaker hydrogen bond acids than phenol itself while 4-nitrophenol and 4-chlorophenol are the strongest hydrogen-bond acids of the para substituted phenols. For the S descriptor the ranking order is –CH3 < –Cl < –Br < –NO2 ≈ –C6H5 < –CN with a range of 0.775 to 1.267 and for the B descriptor –Cl ≈ –Br ≈ < –NO2 < –CH3 < –CN < C6H5– with a range of 0.208 to 0.440. All the para-substituted phenols are more dipolar/polarizable than phenol while 4-chlorophenol and 4-bromophenol are weaker hydrogen-bond bases than phenol with the other para-substituted phenols stronger hydrogen-bond bases. These scales reflect both electronic effects and the intrinsic dipolarity/polarizability and hydrogen-bond basicity of the substituent groups and are not simply related to changes in the property of the phenolic hydroxyl group. However, they provide an indication that variation of the substituent groups has a considerable influence on the overall capability of phenols for intermolecular interactions.
3.3 Anilines and Amides
The contribution of the –NH2 group to the solvation properties of aniline can be estimated as S = 0.50, A = 0.238 and B = 0.28 assuming additivity of the benzene ring and –NH2 group. This is not verified as optimized descriptors for similar aromatic compounds are unavailable. The –NH2 group is a weaker hydrogen-bond acid and stronger hydrogen-bond base than the –OH group in a phenol. An ortho methyl group reduces the hydrogen-bond acidity of aniline, A = 0.163 compared with A = 0.238, but is only slightly more effective than a para methyl group, A = 0.155, Table 7. Electronic effects are likely more important for the monomethyl anilines with a methyl group in any position reducing the hydrogen-bond acidity. All monomethylanilines are slightly stronger hydrogen-bond bases than aniline, B = 0.486 to 0.533 compared with 0.421. The monomethylanilines are about as dipolar/polarizable as aniline itself with an average value of S = 1.041 (0.020, n = 4), Table 7. There is no evidence to suggest that an ortho –Cl or –NO2 group promotes intramolecular hydrogen-bond formation since both 2-chloroaniline and 2-nitroaniline are slightly stronger hydrogen-bond acids than aniline itself, Table 7. This is supported by distance calculations which indicate that the two functional groups are likely too far apart for intramolecular hydrogen-bond formation [54]. The –NO2 group is very effective at enhancing the hydrogen-bond acidity of aniline with all values for the A descriptor significantly greater than for aniline itself. They follow the order 4-nitroaniline > 3-nitroaniline > 2-nitroaniline > aniline with a range of A = 0.622 to 0.238. The same order is observed for the S descriptor. All the mononitroanilines are weaker hydrogen-bond bases than aniline but the range for the B descriptor is small, 0.342 to 0.380, compared with the A descriptor (0.350 to 0.622) and S descriptor (1.456 to 1.804). The introduction of an electron withdrawing group (–NO2) para to –NH2 enhances the S and A descriptors significantly while electron donating groups (–F, –Cl) have a limited effect.
The descriptors for three primary amides with a CH3–, C6H5– and C6H5CH2– group attached to the carbon atom of the amide and one secondary amide with a C6H5– group attached to the nitrogen atom of the amide are given in Table 7. The amide group is more complex than the functional groups considered so far as it contains two potential electron donor sites at the carbonyl oxygen and amine nitrogen [55]. For the three primary amides the principle of additivity is a poor model to assign hydrogen-bonding properties to the amide group. The primary amides are relatively strong hydrogen-bond acids and bases compared with primary alcohols and anilines and occupy the same descriptor space as phenols. They are generally more dipolar/polarizable than typical primary alcohols, phenols and anilines containing alkyl and phenyl substituents. The hydrogen-bond acidity of the primary amides follows the order CH3– > C6H5– > C6H5CH2– with hydrogen-bond basicity following the reverse order. The tighter binding of electron lone pairs in the amide group for the phenyl-containing substituents likely contributes to the observed order. Acetanilide is a secondary amide with a phenyl group attached to the amide nitrogen and a constitutional isomer of 2-phenylacetamide. It is a stronger hydrogen-bond acid than 2-phenylacetamide, A = 0.515 compared with A = 0.436, and a significantly weaker hydrogen-bond base, B = 0.700 compared with B = 0.933. Activation of the amide hydrogen by direct attachment of the phenyl ring to nitrogen must more than compensate for the loss of one acidic hydrogen in the amide group of acetanilide.
3.4 Comparison of Abraham’s Hydrogen-Bonding Descriptors with the Optimized Descriptors
Abraham’s descriptors and the optimized descriptors reported here are distinguished in the remainder of this report by the symbols A(Abr) and A(Opt), respectively, to specify the dataset referred to. The A(Opt) descriptors are calculated using experimental data mostly from a single laboratory utilizing defined experimental protocols and subject to statistical control [13,14,15,16,17,18,19,20,21]. The A(Abr) descriptors are calculated from experimental data from various literature sources and include some data obtained by estimation methods. Thus, differences in the accuracy of the two sets of descriptors can be anticipated [6, 13, 18, 56]. For the alcohols, phenols, anilines, and amides discussed so far (Sects. 3.1 to 3.3) a comparison of the optimized hydrogen-bonding and dipolarity/polarizability descriptors can be made with Abraham’s descriptors taken from [12]. The homologous series of n-alkyl alcohols are treated as a single point since on both scales of hydrogen-bond acidity and basicity they are assigned a constant value independent of the alkyl chain length. A(Opt) is plotted against A(Abr) in Fig. 3. The dispersion of the data points indicates the likely variation of the A descriptor for individual compounds for the two datasets. The two datasets can be fit to a linear model which explains about 92% of the variance
The model intercept includes zero at the 95% confidence level. Both descriptor sets have a common origin, or zero point, and differ mainly in the scale factor with A(Opt) ≈ 1.27 A(Abr). Extreme differences can be noted for some compounds such as 1,12-dodecanediol with A(Opt) = 0.690 and A(Abr) = 0.75, 2-chlorophenol with A(Opt) = 0.520 and A(Abr) = 0.32, and the primary and secondary aliphatic (but not aromatic) alcohols with A(Opt) ≈ 0.9 A(Abr). Apart from these alcohols there are no other specific trends related to compound type. In the solvation parameter model, the contribution of hydrogen-bond acidity to solvation properties is defined by the product term, aA, and so when comparing models based on either dataset, it is important to identify the descriptor database, since the system constants will be smaller in the case of A(Opt) compared with A(Abr).

Plot of the descriptors A(Opt) against A(Abr)
B(Opt) is plotted against B(Abr) in Fig. 4. There is better agreement between the two scales as indicated by the lower dispersion except for 1,12-dodecanediol for which B(Opt) = 1.433 and B(Abr) = 0.99, and is treated as an outlier in the following regression model
The slope for the regression model includes one and the intercept zero at the 95% confidence level. From this we can conclude that the two scales are essentially identical and the disagreement in the B descriptor for individual compounds is largely the results of the variation in the random error in the experimental data used for the descriptor calculations.

Plot of the descriptors B(Opt) against B(Abr). Identification: 1 = 1,12-dodecanediol
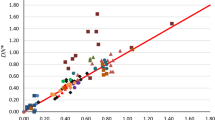
S(Opt) is plotted against S(Abr) in Fig. 5. There is considerable scatter for this plot. Notable is that five out of the eight phenols containing functional groups other than methyl are poorly correlated by the two scales. The phenols with extreme differences for the S descriptor are 2-chlorophenol S(Opt) = 0.676 and S(Abr) = 0.88, 4-chlorophenol S(Opt) = 0.835 and S(Abr) = 1.08, 3-nitrophenol S(Opt) = 1.246 and S(Abr) = 1.57, 4-nitrophenol S(Opt) = 1.193 and S(Abr) = 1.72, and 4-cyanophenol S(Opt) = 1.267 and S(Abr) = 1.63. In each case S(Opt) is less than S(Abr). The remaining phenols conform to the general correlation model for the other hydrogen-bond-capable compounds. For the reduced dataset, the regression model becomes
The slope for the regression model includes 1 and the intercept 0 at the 95% confidence level. However, the predictive capability of the regression model is poor and the need to remove some of the phenols would indicate that the two scales should not be regarded as equivalent.

Plot of the descriptors S(Opt) against S(Abr)
3.5 Construction of an NMR Correlation Model
Abraham et al. have shown that proton NMR can be used to estimate the hydrogen-bond acidity of a compound through a correlation model that relates the difference in chemical shift for a protic hydrogen in the solvents dimethyl sulfoxide (DMSO) and deuterated chloroform (CDCl3), Δδ = δ(DMSO) − δ(CDCl3), to the A(Abr) scale [57].
A unique feature of this approach is that Δδ can be used to determine the hydrogen-bond acidity of individual functional groups in multifunctional compounds if they do not interconvert rapidly on the NMR time scale. Dimerization in CDCl3 and a concentration dependence on the assigned proton chemical shift may limit the accuracy of the method, which is stated to be about 0.05 A(Abr) units [57]. To establish whether a similar relationship exists for A(Opt), a subset of the published chemical shift data [50, 57, 58] that overlapped with the compounds characterized in this report with the addition of N-methylacetamide, 2-methoxyethanol, benzene, styrene, N-methylaniline and diphenylamine to better populate the range for the A(Opt) descriptor was used. For the added compounds the hydrogen-bond acidity was optimized as before giving A(Opt) = 0.185 for N-methylacetamide, 0.356 for 2-methoxyethanol, 0 for benzene and styrene, 0.159 for N-methylaniline, and 0.214 for diphenylamine. The plot of A(Opt) against Δδ is given in Fig. 6 and the data fit to a linear regression model
The compounds contained on the plot are indicated in the legend to Fig. 6. For the same compounds, the plot of A(Abr) against Δδ is shown in Fig. 7 and fit to a linear regression model
There is good agreement for the coefficients of Eqs. 8 and 10 indicating that the subset of data used here has not distorted the original correlation model allowing a meaningful comparison of the A(Opt) and A(Abr) descriptors. Equations 9 and 10 are of similar quality and the NMR method can be used to estimate the A(Opt) descriptor about as well as the A(Abr) descriptor with the appropriate correlation model. Neither acetamide nor benzamide are a good fit to either correlation model and 2-chlorophenol is an extreme value in the A(Abr) model. There is considerable scatter in both plots and the NMR method cannot be used to verify which set of hydrogen-bond acid descriptors is more reliable than the other. The NMR method is probably best considered a useful estimation method for either set of hydrogen-bond acid descriptors with some reservations if used as a standalone determination method.

Plot of A(Opt) against the difference in NMR chemical shift Δδ (see text) for the compounds benzene, styrene, butan-1-ol, 2-methoxyethanol, phenol, 1,3-dihydroxybenzene, 2-chlorophenol, 2-nitrophenol, 4-nitrophenol, 4-cyanophenol, acetamide, N-methylacetamide, benzamide, cyclohexanone N-oxime, aniline, N-methylaniline, diphenylamine, 2-methylaniline, 2-chloroaniline, and 2-nitroaniline

Plot of A(Abr) against the difference in NMR chemical shift Δδ (see text) for the same compounds in Fig. 6
4 Conclusion
The Solver method provides a convenient approach for the calculation of descriptors when large data sets are employed to minimize the impact of experimental error on the descriptor values. It also facilitates the addition of new experimental data when it becomes available as might be needed to improve descriptor estimates for compounds that produce relatively flat descriptor wells. Compared with Abraham’s descriptors the optimized descriptors show mainly a difference in the scale factor for the hydrogen-bond acidity descriptor, A(Opt) ≈ 1.27 A(Abr), and similar values for the hydrogen-bond basicity descriptor, B(Opt) ≈ B(Abr). Dispersion in the correlation models indicate that on a compound-by-compound basis there are general differences in the A and B descriptors other than scaling differences even though the core data is moderately well correlated (A(Abr) can explain > 90% of the variance in A(Opt). The dipolarity/polarizability descriptor S is only poorly correlated for the two scales with systematic differences for phenols with polar ring substituents.
The optimized descriptors were used to explore the effect of targeted structure variations on the hydrogen-bonding properties of hydroxyl groups in alcohols and phenols and amine groups in anilines and amides. The principle of additivity was partially successful for compounds containing only hydrocarbon fragments but was limited for multifunctional compounds in general. It cannot be relied upon as a simple rule for estimating hydrogen-bond descriptors in a general context while for specific examples could prove useful. Compounds containing a phenyl ring often exhibited different behavior to alkyl systems where electronic (inductive and mesomeric), steric, and intramolecular hydrogen-bonding impact hydrogen-bonding descriptors for hydroxyl and amine functional groups. The range of values for these compounds is an indication of the importance of electronic and proximity effects and the likelihood that these effects operate simultaneously presenting difficulties for estimating descriptors by simple models or consideration of 2D-structures.
Data Availability
All experimental data is in the public domain and can be retrieved from the cited sources without hinderance for free.
Code Availability
Not applicable.
References
Abraham, M.H.: Scales of solute hydrogen-bonding: their construction and approach to physicochemical and biochemical processes. Chem. Soc. Rev. 22, 73–83 (1993)
Abraham, M.H., Platts, J.A.: Hydrogen bond structural group constants. J. Org. Chem. 66, 3484–3491 (2001)
Abraham, M.H., Gola, J.M.R., Cometto-Muniz, J.E., Acree, W.E.: Hydrogen bonding between solutes in solvents octan-1-ol and water. J. Org. Chem. 75, 7651–7658 (2010)
Abraham, M.H., Poole, C.F., Poole, S.K.: Classification of stationary phases and other materials by gas chromatography. J. Chromatogr. A 842, 79–114 (1999)
Abraham, M.H., Ibrahim, A., Zissimos, A.M.: Determination of sets of solute descriptors from chromatographic measurements. J. Chromatogr. A 1037, 29–47 (2004)
Poole, C.F., Ariyasena, T.C., Lenca, N.: Estimation of the environmental properties of compounds from chromatographic measurements and the solvation parameter model. J. Chromatogr. A 1317, 85–104 (2013)
Abraham, M.H., Whiting, G.S., Doherty, R.M., Shuely, W.J.: Hydrogen bonding 13. A new method for the characterization of GC stationary phases—the Laffort data set. J. Chem. Soc. Perkin Trans. 2, 1451–1460 (1990)
Abraham, M.H., Whitting, G.S., Doherty, R.M., Shuely, W.J.: Hydrogen-bonding 16: a new solute solvation parameter, π2H, from gas chromatographic data. J. Chromatogr. 587, 213–228 (1991)
Martin, S.D., Poole, C.F., Abraham, M.H.: Synthesis and gas chromatographic evaluation of a high-temperature hydrogen-bond acid stationary phase. J. Chromatogr. A 805, 217–235 (1998)
Abraham, M.H.: Hydrogen-bonding 31: construction of a scale of solute effective or summation hydrogen-bond basicity. J. Phys. Org. Chem. 6, 660–684 (1993)
ADME suite 5.0, Advanced Chemistry Development, Toronto, ON.
Ulrich, N., Endo, S., Brown, T.N., Watanabe, N., Bronner, C., Abraham, M.H., Goss, K.-H.: UFZ-LSER Database v. 3.2.1 [internet]. Helmholtz Centre for Environmental Research-UFZ, Leipzig (2017)
Poole, C.F.: Wayne State University experimental descriptor database for use with the solvation parameter model. J. Chromatogr. A 1617, 460841 (2020)
Poole, C.F.: Selection of calibration compounds for selectivity evaluation of wall-coated, open-tubular columns for gas chromatography by the solvation parameter model. J. Chromatogr. A 1629, 461500 (2020)
Poole, C.F.: Selection of calibration compounds for selectivity evaluation of siloxane-bonded silica columns for reversed-phase liquid chromatography by the solvation parameter model. J. Chromatogr. A 1633, 461652 (2020)
Poole, C.F., Atapattu, S.N.: Selectivity evaluation of core-shell silica columns for reversed-phase liquid chromatography using the solvation parameter model. J. Chromatogr. A 1634, 461692 (2020)
Poole, C.F.: Evaluation of the solvation parameter model as a quantitative structure-retention relationship model for gas and liquid chromatography. J. Chromatogr. A 1626, 461308 (2020)
Poole, C.F., Atapattu, S.N., Bell, A.K.: Determination of solute descriptors by chromatographic methods. Anal. Chim. Acta 652, 32–53 (2009)
Poole, C.F., Ahmed, H., Kiridena, W., Patchett, C.C., Koziol, W.W.: Revised solute descriptors for characterizing retention properties of open-tubular columns in gas chromatography and their application to a carborane-siloxane copolymer stationary phase. J. Chromatogr. A 1104, 299–312 (2006)
Poole, C.F.: Solvation parameter model: tutorial on its application to separation systems for neutral compounds. J. Chromatogr. A 1645, 462108 (2021)
Poole, C.F.: Gas chromatography system constant database for 52 wall-coated, open-tubular columns covering the temperature range 60–140 °C. J. Chromatogr. A 1604, 460482 (2019)
Poole, C.F.: Gas chromatography system constant database over an extended temperature range for nine open-tubular columns. J. Chromatogr. A 1590, 130–145 (2019)
Lenca, N., Poole, C.F.: A system map for the ionic liquid stationary phase 1,9-Di(3-vinylimidazolium)nonane Bis(trifluoromethylsulfonyl)imide. Chromatographia 78, 81–88 (2015)
Lenca, N., Poole, C.F.: A system map for the ionic liquid stationary phase tri(tripropylphosphoniumhexanamido)triethylamine bis(trifluoromethylsulfonyl)imide for gas chromatography. J. Chromatogr. A 1524, 210–214 (2017)
Lenca, N., Poole, C.F.: A system maps for the ionic liquid stationary phase 1,12-di(tripropylphosphonium)dodecane bis(trifluoromethylsulfonyl)imide for gas chromatography. J. Chromatogr. A 1525, 138–144 (2017)
Lenca, N., Poole, C.F.: A system maps for the ionic liquid stationary phase 1,12-di(tripropylphosphonium)dodecane bis(trifluoromethylsulfonyl)imide trifluoromethanesulfonate for gas chromatography. J. Chromatogr. A 1559, 164–169 (2018)
Abraham, M.H., Andonian-Haftvan, J., Hamerton, I., Poole, C.F., Kollie, T.O.: Comparison of the solvation theories of Abraham and Poole, using a new acidic GLC stationary phase. J. Chromatogr. 646, 351–360 (1993)
Poole, C.F.: Reversed-phase liquid chromatography system constant database over an extended mobile phase composition range for 25 siloxane-bonded silica-based columns. J. Chromatogr. A 1600, 112–126 (2019)
Atapattu, S.N.: Solvation properties of acetone–water mobile phases in reversed-phase liquid chromatography. J. Chromatogr. A 1650, 462252 (2021)
Poole, C.F.: Partition constant database for totally organic biphasic systems. J. Chromatogr. A 1527, 18–32 (2017)
Ariyasena, T.C., Poole, C.F.: Models for liquid–liquid partition in the system ethanolamine–organic solvent and their use for estimating descriptors for organic compounds. Chromatographia 76, 157–164 (2013)
Ariyasena, T.C., Poole, C.F.: Evaluation of triethylamine as a counter solvent in totally organic biphasic liquid–liquid partition systems. Chromatographia 76, 1031–1039 (2013)
Hansch, C., Leo, A., Hoekman, D.: Q.S.A.R. Exploring Hydrophobic, Electronic, and Steric Constraints. American Chemical Society, Washington, DC (1995)
Abraham, M.H., Platts, J.A., Hersey, A., Leo, A.J., Taft, R.W.: Correlation and estimation of gas–chloroform and water–chloroform partition coefficients by a linear free energy relationship method. J. Pharm. Sci. 88, 670–679 (1999)
Sprunger, L.M., Gibbs, J., Acree, W.E., Abraham, M.H.: Correlation and prediction of partition coefficients for solute transfer to 1,2-dichloroethane from both water and from the gas phase. Fluid Phase Equilibr. 273, 78–86 (2008)
Qian, J., Poole, C.F.: Distribution model for Folch partition. J. Sep. Sci. 30, 2326–2331 (2007)
Abraham, M.H., Chadha, H.S., Whiting, G.S., Mitchell, R.C.: Hydrogen bonding 32. An analysis of water–octanol and water–alkane partitioning and the Δlog P parameter of Seller. J. Pharm. Sci. 83, 1085–1100 (1994)
Platts, J.A., Butina, D., Abraham, M.H., Hersey, A.: Estimation of molecular linear free energy relation descriptors using a group contribution approach. J. Chem. Inf. Comput. Sci. 39, 825–845 (1999)
Ariyasena, T.C., Poole, C.F.: Determination of descriptors for polycyclic aromatic hydrocarbons and related compounds by chromatographic methods and liquid–liquid partition in totally organic biphasic systems. J. Chromatogr. A 1361, 240–254 (2014)
Sheldon, T.J., Adjiman, C.S., Cordiner, J.L.: Pure component properties from group contribution: hydrogen-bond basicity, Hildebrand solubility parameter, macroscopic surface tension, dipole moment, refractive index and dielectric constant. Fluid Phase Equilibr. 231, 27–37 (2005)
Schuurmann, G., Ebert, R.-U., Kuehne, R.: Prediction of physicochemical properties of organic compounds from 2D molecular structure—fragment methods vs. LFER models. Chimia 60, 691–698 (2006)
Cacelli, I., Campanile, S., Giolitt, A., Molin, D.: Theoretical prediction of the Abraham hydrogen bond acidity and basicity factors from a reaction field method. J. Chem. Inf. Model. 45, 327–333 (2005)
Devereux, M., Popelier, P.L.A., Mclay, I.M.: A refined model for prediction of hydrogen bond acidity and basicity parameters from quantum chemical molecular descriptors. Phys. Chem. Chem. Phys. 11, 1595–1603 (2009)
Schwobel, J.A.H., Ebert, R.-U., Kuhne, R., Schuurmann, G.: Prediction models for Abraham hydrogen bond donor strength: comparison of semi-empirical, ab initio, and DFT methods. J. Phys. Org. Chem. 24, 1072–1080 (2011)
Rahaman, O., Doren, D.J., Di Toro, D.M.: Quantum mechanical estimation of Abraham hydrogen bond parameters using 1:1 donor-acceptor complexes. J. Phys. Org. Chem. 27, 783–793 (2014)
Liang, Y.-Z., Xiong, R.C., Sandler, S.I., Di Toro, D.M.: Quantum chemically estimated Abraham solute parameters using multiple solvent–water partition coefficients and molecular polarizability. Environ. Sci. Technol. 51, 9887–9898 (2017)
Lenca, N., Atapattu, S.N., Poole, C.F.: Estimation of descriptors for hydrogen-bonding compounds from chromatographic and liquid–liquid partition measurements. J. Chromatogr. A 1526, 13–22 (2017)
Abraham, M.H., Du, C.M., Platts, J.A.: Lipophilicity of nitrophenols. J. Org. Chem. 65, 7114–7118 (2000)
Abraham, M.H., Dearden, J.C., Bresnen, G.M.: Hydrogen bonding steric effects and thermodynamics of partitioning. J. Phys. Org. Chem. 19, 242–248 (2006)
Abraham, M.H., Abraham, R.J., Acree, W.E., Aliev, A.E., Leo, A., Whaley, W.L.: An NMR method for the quantitative assessment of intermolecular hydrogen bonding: application to physicochemical, environmental, and biochemical properties. J. Org. Chem. 79, 11075–11083 (2014)
Abraham, M.H., Abraham, R.J., Aliev, A.E., Tormena, C.F.: Is there an intramolecular bond in 2-halophenols? A theoretical and spectroscopic investigation. Phys. Chem. Chem. Phys. 17, 25151–25159 (2015)
Abraham, M.H., Acree, W.E., Earp, C.E., Vladimirova, A., Whaley, W.L.: Studies on the hydrogen bond acidity and other descriptors and properties for hydroxyflavones and hydroxyisoflavones. J. Mol. Liq. 208, 363–372 (2015)
Acree, W.E., Smart, K., Abraham, M.H.: Abraham model solute descriptors reveal strong intramolecular hydrogen bonding in 1,4-dihydroxyanthraquinone and 1,8-dihydroxyanthraquinone. Phys. Chem. Liq. 56, 416–420 (2018)
Abraham, M.H., Abraham, R.J., Aghamohammadi, A., Afarinkia, K., Liu, X.L.: The assessment of intramolecular hydrogen bonding in ortho-substituted anilines by an NMR method. J. Mol. Liq. 315, 113730 (2020)
Abraham, M.H., Abraham, R.J.: A simple and facile NMR method for the determination of hydrogen bonding by amide N-H protons in protein models and other compounds. New J. Chem. 41, 6064–6066 (2017)
Atapattu, S.N., Poole, C.F.: Solute descriptors for characterizing retention properties of open-tubular columns of different selectivity in gas chromatography at intermediate temperatures. J. Chromatogr. A 1195, 136–145 (2008)
Abraham, M.H., Abraham, R.J., Byrne, J., Griffiths, L.: NMR method for the determination of solute hydrogen bond acidity. J. Org. Chem. 71, 3389–3394 (2006)
Abraham, M.H., Gil-Lostes, J., Cometto-Muniz, J.E., Cain, W.S., Poole, C.F., Atapattu, S.N., Abraham, R.J., Leonard, P.: The hydrogen-bond acidity and other descriptors for oximes. New J. Chem. 33, 76–81 (2009)
Funding
No funding was received for conducting this study.
Author information
Authors and Affiliations
Contributions
CFP is the sole author of this article and responsible for all elements of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The author has no financial or proprietary interest in any of the material discussed in this article.
Ethical Approval
Not applicable. No human or animal data used in writing this manuscript.
Consent to Participate
Not applicable.
Consent for Publication
Wayne State University agrees to the submission of this article by the author.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Poole, C.F. Structural Effects on the Hydrogen-Bonding Descriptors of the Solvation Parameter Model. J Solution Chem 51, 1056–1080 (2022). https://doi.org/10.1007/s10953-021-01133-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10953-021-01133-z