
Abstract
A thorough search of the scientific literature under the auspices of the IUPAC Sub-Committee on Solubility and Equilibrium Data (SSED) has identified and compiled quantitative thermodynamic data for the first sulfate protonation step from about 270 papers, reports, books and electronic databases. A critical evaluation of these sources using well-defined criteria has rejected about half of them. The remaining (‘accepted’) data reveal that the standard state values of the first protonation constant of the sulfate ion, \(K_{1}^{\text{o}}\), corresponding to the equilibrium: \({\text{SO}_{4}^{2 - }} ({\text{aq}}) + {\text{ H}}^{ + } \left( {\text{aq}} \right) \rightleftharpoons {{\text{HSO}}_{4}^{ - }}({\text{aq}})\) at infinite dilution, are known to good levels of accuracy up to ~ 250 °C. However, at higher temperatures, and at all temperatures in the presence of added electrolytes, the equilibrium constant values are much less certain. The corresponding values for the enthalpy (\(\Delta_{\text{r}} H_{1}^{\text{o}}\)), entropy (\(\Delta_{\text{r}} S_{1}^{\text{o}}\)) and isobaric heat capacity (\(\Delta_{\text{r}} C_{p,1}^{\text{o}}\)) changes are also moderately well determined at near-ambient temperatures but are much more poorly defined both at higher temperatures and in the presence of even modest concentrations of added electrolytes. Comments on a number of aspects of the data and their evaluation are provided.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Background
1.1 The Importance of Sulfuric Acid and the Sulfate Ion
Solutions containing sulfuric acid and/or sulfate ions (‘acidic sulfate solutions’) are of unsurpassed importance industrially and throughout the natural environment [1,2,3]. In aqueous solution the two species are linked by the following protonation equilibria:
In industrial terms the production of sulfuric acid dwarfs those of all other anthropogenic chemicals, with a current (and increasing) global output of ~ 260 Mt·year−1, which is about twice that of the next largest product. So critical is sulfuric acid production in modern economies that it has been used as a comparative measure of national industrial productivity [1].
About 50% of world output of sulfuric acid is used in the manufacture of the phosphate fertilizers, which are essential for global food production. Many key metals, such as Cu, Zn and Li, are extracted and/or purified using acidic sulfate solutions [2, 4]. So-called ‘lead–acid’ batteries are still produced in far greater numbers than any other rechargeable power source, despite the recent advances in alternative battery types [5]. In addition to these large-scale applications, acidic sulfate solutions are used for a myriad of purposes, e.g., in paints, pigments and dyes, oil refining and so on [1, 2].
One area where the interplay between sulfuric acid and the sulfate ion is especially important, for both economic and environmental reasons, is in acid mine drainage and the development of acid sulfate soils [6]. Depending on the nature of the partnering cation, sulfate salts also have an almost unlimited range of applications that are far too numerous to list here. Equally important, the sulfate anion is ubiquitous throughout the human and physical environments [1, 3]. Typical concentrations of sulfate in a variety of bodily and environmental fluids are listed in Table 1.
This panorama of applications and implications underlines the need for reliable quantitative knowledge of the behavior of acidic sulfate solutions over wide ranges of conditions.
1.2 The Nature of Sulfuric Acid Solutions
Elementary chemistry textbooks usually describe sulfuric acid as being a strong diprotic acid in aqueous solution [7]. That is, both equilibria (1) and (2) lie fully to the left (sulfuric acid is said to be ‘fully dissociated’). This description is indeed apt for dilute solutions of sulfuric acid in water. For example, the overall dissociation reaction:
is > 90% complete for a 1 mmol·L−1 sulfuric acid solution at near-ambient temperatures (where, here and throughout, mol·L−1 ≡ mol·dm−3). And of course, in analytical terms, a solution of sulfuric acid titrates straightforwardly with any strong base, e.g., NaOH(aq), as a diprotic acid [8].
Nevertheless, as has been well documented for more than a century [9, 10], on the basis of electrical conductivity, potentiometry and other measurements, there is no doubt that sulfuric acid is not fully dissociated at most concentrations in aqueous solution. Indeed, in many situations it seems best to describe sulfuric acid as a strong monoprotic acid:
which corresponds to equilibrium (2) lying fully to the left while equilibrium (1) does not.
At high concentrations, say > 5 mol·dm−3, the nature of sulfuric acid solutions undergoes a transformation, becoming more molecular/less ionic, causing the chemical speciation to become much more complicated [1]. There is an extensive literature on the nature and relative abundances of the various species in highly concentrated sulfuric acid; for example, Greenwood and Earnshaw list six species that constitute about 0.4 mol% of pure (liquid) H2SO4 [1]. However, because such concentrations of H2SO4 are far above those that are typically encountered in the environment or used industrially, they will not be considered further in this review.
1.3 The Importance of Thermodynamic Data for Acidic Sulfate Solutions
Because of the widespread occurrence and use of aqueous solutions of sulfuric acid, with or without the presence of metal sulfates, vast numbers of quantitative and qualitative studies (in excess of one thousand) containing thermodynamic data for sulfate species have appeared in the literature [11]. These data have been used extensively for the interpretation of a variety of chemical phenomena and are particularly important for inclusion in the computerized databases that inform chemical speciation modelling programs. Such programs are used commonly nowadays for scientific and environmental purposes, industrial process optimization and control, in the treatment and storage of nuclear waste, and for modelling geochemical processes under widely varying conditions.
As for many equilibria, quantitative data for the protonation of the sulfate ion are spread widely throughout the scientific literature, frequently in obscure journals or in unpublished reports but, unfortunately, are often of dubious reliability. These factors make identification of accurate data difficult for the non-expert and there are many instances where unreliable stability constants (or related thermodynamic parameters) have become embedded in the literature and databases and used uncritically.
The importance of reliable thermodynamic data for sulfate equilibria in solution has been recognized by the Sub-Committee on Solubility and Equilibrium Data (SSED) of the International Union of Pure & Applied Chemistry (IUPAC). This body has established a project, Stability Constants and Related Thermodynamic Data of Metal-Ion/Sulfate Complexes in Aqueous Solution, which is being undertaken by the present authors [11]. This IUPAC-sponsored project aims to compile all the existing data, critically evaluate them, identify unreliable data, and make recommendations regarding the current ‘best-available’ (hopefully most-accurate) values. These values will cover as wide ranges as possible with respect to temperature, pressure, ionic strength I, and composition (medium).
The present paper summarizes some of the authors’ recent experiences in collecting and assessing thermodynamic data relevant to the first protonation of the sulfate ion, equilibrium (1). This is undoubtedly the most important reaction/equilibrium occurring in most acidic sulfate solutions, and one of the most thoroughly investigated in all chemistry. In keeping with the long-standing practice of the SSED and its predecessor bodies {the former IUPAC Commissions V.8 (Solubility Data) and V.6 (Stability Constants)} only experimental data are considered. The idea behind this approach is that theoreticians and computer modellers can then use the most reliable data available to develop or test their theories and models free from bias.
It is emphasised that the present paper describes ongoing work. The numerical values reported here are still ‘fluid’ and will certainly change (but probably only slightly) as the assessment of the data is further refined. The ultimate compilation and findings of this work will be published, subject to approval by the SSED and IUPAC, in the journal, Pure and Applied Chemistry.
2 The First Protonation Constant of the Sulfate Ion
As already noted, at most concentrations of practical importance, equilibrium (2) lies completely to the left and only equilibrium (1) needs to be considered. The equilibrium constant, K1, for the first protonation (acid association) of the sulfate ion can be written in concentration terms (omitting the ‘aq’ state symbols) as:
A similar expression in terms of molalities can be written for measurements made using mol·kg−1 concentrations but note that the numerical values of K1 will differ [12].
The standard state (infinite dilution in water) constant, \(K_{1}^{\text{o}}\), is properly expressed in terms of activities, a:
where a(i) = c(i)·f(i), with c(i) and f(i) being respectively the concentration and activity coefficient (on the molarity scale) of the species i. Values of \(K_{1}^{\text{o}}\) cannot be measured experimentally because they involve single ion activities [14]. Instead, they must be estimated, either by extrapolation to infinite dilution of K1 values measured at finite concentrations or by conversion of such values to infinite dilution using an appropriate (non-thermodynamic) activity coefficient equation [14]. It may be noted that the difference between these two approaches is largely semantic since the same theories are used for both extrapolation and calculation. It should also be noted that K1 depends on the ionic strength I and the nature of the medium, while \(K_{1}^{\text{o}}\) does not; both constants depend on temperature and pressure.
2.1 Database
The number of studies reporting quantitative data for the first protonation constant of the sulfate ion is almost overwhelming. Approximately 270 papers, reports and books were located [11] that contained numerical values of K1 and/or \(K_{1}^{\text{o}}\). The earliest quantitative values reported for K1 appear to be those of Luther [15] and of Noyes and Eastman [9]. These authors calculated K1 and the enthalpy change of reaction (1) using freezing point, conductivity and thermochemical data from earlier studies by other investigators. Some of the early estimates are associated with very well-known scientists of their day: Kohlrausch in Germany [16] and Noyes in the USA [9, 17]. Staples [10] has made a careful study of these early results (in terms of activity coefficients) and found many to be consistent with modern-day estimates, even including some that pre-date the 1923 Debye–Hückel theory, which provided the first theoretically rigorous basis for interpreting the behavior of dilute electrolyte solutions [14].
After more than 100 years of quantitative investigations, interest in sulfate protonation persists, with an ongoing (albeit modest) stream of papers concerned, in part or whole, with the thermodynamic characteristics of equilibrium (1). Recent focus has largely been on development of computer speciation models, mostly based on Pitzer equations, of very complex mixtures of acidic sulfate solutions over wide ranges of temperature relevant to hydrometallurgical or environmental processes [18,19,20].
Of the ca. 270 reports identified [11] as containing quantitative thermodynamic information for equilibrium (1), approximately 50% of the stated numerical values were rejected outright and excluded from the subsequent critical evaluation. These rejections were based on well-established criteria employed in previous IUPAC-sponsored critical evaluation exercises [12]. They include: experimental problems such as poorly- or un-defined conditions (temperature, medium, etc.), use of an inappropriate experimental method, and inadequate or incorrect processing of results. As only experimental results were to be included, all values predicted from theories or by computer modelling alone were rejected: these approaches are far too primitive at their present levels of development to provide reliable estimates of thermodynamic parameters in solutions. Another group of values rejected were those that were merely uncritical repetitions of literature values, or those given in databases. The latter are an interesting subset: such values are often provided without (or with incorrect) references to the original data, use incorrect or discredited data, or are given with absurd levels of precision that convey an incorrect message about the real uncertainties. Exceptions to this procedure were made for recalculations of high quality experimental data using a significantly improved theory, or for the correction of errors (with appropriate justification).
After this initial selection, a ‘democracy of observers’ was followed, i.e., all accepted values were treated equally regardless of their source. A partial exception to this approach was made for multiple publications from the same (or a closely related) source, since it seems to be a fact of science that individual laboratories or groups have much less difficulty reproducing their own results than reproducing those of others. To avoid over-weighting, such values were usually combined into a single average result. And inevitably, while democracy is a worthy ideal, it was necessary on a few occasions to adapt George Orwell’s [21] ironic dictum: “All [scientists] are equal, but some are more equal than others” when trying to reconcile disparate results among independent workers.
2.2 Value of \(\log_{10} K_{1}^{\text{o}}\) at 25 °C
Although, as already noted, the standard state equilibrium constant for the first protonation of the sulfate ion, \(K_{1}^{\text{o}}\), is not an experimental quantity, it is derived directly from experimental data and is of singular importance [14]. Standard state constants have a very clear interpretation: they are a measure of the position of an equilibrium in the (virtual) absence of all species except the solvent (but including those participating in the equilibrium). They are also the most widely tabulated and cited constants and, very importantly, serve as an anchor to fit data obtained at finite concentrations [14]. As such they provide a means to assess theories (equations) for stability constants and activity coefficients [13].
While the choice of standard state for equilibria in aqueous solution (infinite dilution in water of all dissolved species) is generally agreed upon, the choice of standard conditions is arbitrary. Nevertheless, most values of \(K_{1}^{\text{o}}\) have been reported at 25 °C and 1 atm pressure. Fortunately, the difference between 1 atm (101,325 Pa) and the current IUPAC preference for 1 bar (100,000 Pa) as the standard pressure is insignificant for aqueous solutions at near-ambient temperatures.
Almost 130 experimental estimates of \(K_{1}^{\text{o}}\) at 25 °C were found in the literature [11], which probably makes equilibrium (1) the second-most intensively studied of all solution equilibria (the self-ionization of water being first [22, 23]). About 85 of these estimates were rejected leaving 43 values on which to base further evaluation. The unweighted average of these estimates gave: \(\log_{10} K_{1}^{\text{o}}\) = 1.976 ± 0.025 (1 σ), with a range 1.92 ≤ \(\log_{10} K_{1}^{\text{o}}\) ≤ 2.02 and a mode (most commonly reported value) of \(\log_{10} K_{1}^{\text{o}}\) = 1.99. An interesting feature of the accepted data is revealed in Fig. 1, namely that the distribution of the accepted \(\log_{10} K_{1}^{\text{o}}\) values is asymmetric. Further examination revealed that most of the lower values came from papers published prior to 1963, with most appearing before 1951. While age of publication was not a routine criterion for rejection (except for most pre-1923 data) it cannot be ignored totally and further consideration of all the accepted data may well result in rejection of these values [11]. Application of a 1σ rejection criterion to the accepted values gave a final result of \(\log_{10} K_{1}^{\text{o}}\) = 1.984 ± 0.013 (1 σ), with the number of independent values, N = 38. Pending further consideration, this estimate of \(\log_{10} K_{1}^{\text{o}}\) is recommended as the present best-available value.

Distribution diagram for the accepted values of \(\log_{10} K_{1}^{\text{o}}\) at 25 °C
This value for \(\log_{10} K_{1}^{\text{o}}\) corresponds to \(K_{1}^{\text{o}}\) = 96.4 ± 2.9 L·mol−1, i.e., the 1 σ uncertainty in \(K_{1}^{\text{o}}\) at 25 °C is ± 3%. While this may seem large, to give some context, the current best estimate of the self-ionization constant of water at 25 °C is \(K_{\text{w}}^{\text{o}}\) = (1.011 ± 0.011) × 10−14 corresponding to ± 1% uncertainty [23, 24]. It is notable that some authorities [25, 26] recommend the same value for \(K_{1}^{\text{o}}\) but claim ± 1% uncertainty. This lower uncertainty can only be achieved by a more narrow selection of the data.
One last comment on the results for \(\log_{10} K_{1}^{\text{o}}\) at 25 °C is appropriate. The 38 values finally accepted were obtained with a very pleasing spread of techniques: electrical conductivity (4 studies); kinetic measurements (3 studies); solubility measurements (PbSO4 (1 study); CaSO4 (1 study); Ag2SO4 (1 study); 3 studies in all); UV–Vis spectrophotometry (3 studies); Raman spectrometry (4 studies); dilatometry (2 studies) and potentiometry (using quinhydrone (3); glass (4); hydrogen (12) electrodes; 19 studies in all}. This diversity of techniques adds considerable confidence to the recommended result.
2.3 Values of \(\log_{10} K_{1}^{\text{o}}\) as a Function of Temperature
The literature values of \(\log_{10} K_{1}^{\text{o}}\) are plotted in Fig. 2 as a function of temperature. The included data were restricted to temperatures below the critical point of water to avoid any complications due to critical solution phenomena. Note too that Fig. 2 includes some rejected data as well as the accepted values, with only the latter being used to determine recommended values.

Plot of \(\log_{10} K_{1}^{\text{o}}\) as a function of temperature. Note that some rejected (but not all accepted) data are included; references for the cited values are provided in the supplementary material
There are a number of features of the data in Fig. 2 that warrant comment. First, \(\log_{10} K_{1}^{\text{o}}\) is found to increase spectacularly (but more or less smoothly) from ~ 1.7 to ~ 7.5 as the temperature increases over the range 0 ≤ T/°C ≤ 350. Second, the results cluster together reasonably well around the average values up to about 200 °C but diverge increasingly as T increases.
This is certainly a reflection of the increased experimental difficulties at such temperatures (and pressures). Finally, some of the (rejected) results deviate markedly (sometimes by many orders of magnitude in \(K_{1}^{\text{o}}\)) from the bulk of the data. Such results are surprising given that this is a relatively simple system to study (one dominant equilibrium, with a stability constant neither too large nor too small for easy measurement, no side reactions in water, and no redox behavior).
The unweighted averages of the accepted \(\log_{10} K_{1}^{\text{o}} (T)\) values are plotted in Fig. 3 over the temperature range 0 ≤ T/°C ≤ 300. No data were found for T < 0 °C, while the data at T > 300 °C were not considered to be sufficiently reliable to be included. As \(\log_{10} K_{1}^{\text{o}} (T)\) is expected to follow a smooth (albeit not necessarily monotonic) curve, the deviations at T = 5 and 45 °C are almost certainly artefacts due to the limited number of independent reports (1 ≤ N ≤ 6). On the other hand, although it is hard to discern, there is a slight inflexion in \(\log_{10} K_{1}^{\text{o}} (T)\) at T ≳100 °C. The origin of this feature is unknown but a similar inflexion occurs at approximately the same temperature for \({\text{p}}K_{{\text{w}}}^{\text{o}} (T)\); see Sect. 3.3 for further discussion.

Plot of unweighted mean values of \(\log_{10} K_{1}^{\text{o}}\) as a function of temperature
2.4 Variation of log10K 1 with Ionic Strength
Given its widespread application to ‘real’ chemical systems where the presence of other dissolved electrolytes is common and the ionic strength is often quite high, it is surprising to find that there are relatively few published data on the effects of added electrolytes on log10K1, even at 25 °C. At other temperatures and especially at high ionic strengths, such data are almost non-existent, with the notable exception of an outstanding paper by Dickson et al. [27] in NaCl media. Partial exceptions from this unsatisfactory situation are the K1 values reported for NaCl and NaClO4 media at 25 °C [11]. The averages of the available log10K1 data in both media are plotted as a function of ionic strength in Fig. 4.

Plot of averaged log10K1 values at 25 °C as a function of ionic strength, I, in aqueous solutions containing: NaCl (filled circles) and NaClO4 (filled squares) as the swamping electrolyte; (filled star) indicates the infinite dilution value, \(\log_{10} K_{1}^{\text{o}}\)
It should be noted that the average values in NaCl(aq) media are smoother than those in NaClO4(aq) because the former are derived from just a few studies. In contrast, the NaClO4(aq) data are averages of > 20 independent studies [11]; however, the result in 5 mol·L−1 (NaClO4) is from a single study [28] and is almost certainly in error. The two major conclusions to be drawn from Fig. 4 are that the values of log10K1 depend strongly on I but are largely independent of the nature of the medium. Although this observation is based on data for just two added electrolytes (with the same cation), it is still gratifying because such behavior forms the basis of the ionic medium principle that underpins most stability constant measurements and chemical speciation modelling [12, 13]. The nature of the dependence on I in Fig. 4 has been confirmed by isopiestic studies of H2SO4–NaCl solutions [29].
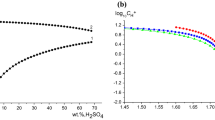
A partial exception to this happy situation is shown in Fig. 5, which plots log10K1 in sulfuric acid media [11]. These values are particularly important for modelling the behavior of hydrometallurgical and other industrial processes that employ moderately concentrated sulfuric acid solutions, with or without metal sulfate salts present. Such measurements are not normally practicable but are feasible in H+/\({\text{HSO}_{4}^{ - }}\)/\({\text{SO}_{4}^{ 2- }}\) solutions because both \({\text{HSO}_{4}^{ - }}\) and \({\text{SO}_{4}^{2 - }}\) have unique Raman and IR signals [30]. Although the data in Fig. 5 are somewhat scattered, they again show that log10K1 depends mostly on I. However, as the sulfuric acid concentration increases there appears to be a significant medium effect, with log10K1 in H2SO4 solutions being considerably smaller than those in NaCl or NaClO4 media (compare Figs. 4 and 5).

Plot of log10K1 values at 25 °C as a function of the stoichiometric concentration of sulfuric acid solutions; references for the cited values are provided in the supplementary material
3 Enthalpy, Entropy and Heat Capacity Changes
3.1 Enthalpy Change
The available literature data for the standard enthalpy change, \(\Delta_{r} H_{1}^{o}\), for the reaction [cf. equilibrium (1)]:
are plotted as a function of temperature in Fig. 6. These values [11] were obtained using various types of calorimetry and from the temperature derivative of \(\log_{10} K_{1}^{\text{o}} (T)\). As for other solution equilibria, there are many fewer values for \(\Delta_{\text{r}} H_{1}^{\text{o}}\) (Fig. 6 includes both accepted and rejected data) than for \(\log_{10} K_{1}^{\text{o}}\) (Fig. 2). This is particularly so at T > 200 °C. At lower T the scatter in the reported values of \(\Delta_{\text{r}} H_{1}^{\text{o}} (T)\) is relatively small, similar to that observed for \(\log_{10} K_{1}^{\text{o}} (T)\).

Enthalpy change for reaction (7) as a function of temperature. Note that some rejected (but not all accepted) data are included; references for the cited values are provided in the supplementary material
As expected from the increase in \(K_{1}^{\text{o}}\) with T (Fig. 2), \(\Delta_{\text{r}} H_{1}^{\text{o}}\) is strongly positive (unfavorable) becoming increasingly so with increasing T (Fig. 6). Given that heat changes accompanying bond formation are usually favorable, this may seem surprising; it results from the large decrease in ion hydration associated with partial charge neutralization (Eq. 7), i.e., with unfavorable ion–water bond breaking [31]. It is noteworthy that the slight inflection in the \(\log_{10} K_{1}^{\text{o}} (T)\) curve at T ≈ 100 °C (Fig. 2) is somewhat more prominent in the \(\Delta_{\text{r}} H_{1}^{\text{o}} (T)\) plot (Fig. 6).
At 25 °C the relatively abundant data give a value of \(\Delta_{\text{r}} H_{1}^{\text{o}}\) = 21.4 ± 0.8 kJ·mol−1 [11]. The 1 σ uncertainty corresponds to ± 4%, which is probably as good as can be expected. There does not appear to be a recent estimate of the uncertainty in the corresponding value for the ionization of water, \(\Delta_{\text{r}} H_{\text{w}}^{\text{o}}\). However, Brown and Ekberg [23] cite an ‘experimental’ value of 55.81 kJ·mol−1 at 25 °C while differentiation of the IAPWS values of \({\text{p}}K_{\text{w}}^{\text{o}} (T)\) yields \(\Delta_{\text{r}} H_{\text{w}}^{\text{o}}\) = 56.38 kJ·mol−1, a difference of ~ 1% [23, 24].
3.2 Entropy Change
The values reported in the literature for the standard entropy change, \(\Delta_{\text{r}} S_{\text{w}}^{\text{o}}\), accompanying reaction (7) are given as a function of temperature in Fig. 7. Almost all of these values were obtained [11] from stability constant and enthalpy change data using the usual thermodynamic relationships: ΔG° = − RT ln K° = ΔH − TΔS°. In this sense, the entropy values do not contain any new information and the commentary that can be made on them is essentially the same as has been made above on \(\Delta_{\text{r}} H_{1}^{\text{o}} .\) The important difference is that the entropy change is strongly positive (favorable) and more than compensates the unfavorable enthalpy change. The origin of this effect is the release of water molecules from the hydration shells of the ions due to partial charge neutralization [31]. At 25 °C the available data average to: \(\Delta_{\text{r}} S_{1}^{\text{o}}\) = 111 ± 3 J·K−1·mol−1, with the 1 σ uncertainty corresponding to ± 3%; of course the maximum uncertainty is higher because ΔS depends on both ΔG and ΔH.

Entropy change for reaction (7) as a function of temperature. Note that some rejected (but not all accepted) data are included; references for the cited values are provided in the supplementary material
3.3 Heat Capacity Change
The rather limited amount of data reported in the literature [11] for \(\Delta_{\text{r}} C_{p,1}^{\text{o}}\), the standard isobaric heat capacity change accompanying reaction (7), is given as a function of temperature in Fig. 8. These values were obtained in a variety of ways: from the second derivative of \(\log_{10} K_{1}^{\text{o}}\) with respect to temperature, from the first temperature derivative of \(\Delta_{\text{r}} H_{1}^{\text{o}}\), and by direct calorimetry [11].

Isobaric heat capacity change for reaction (7) as a function of temperature. Note that some rejected data are included; references for the cited values are provided in the supplementary material
While there are major differences among the independent determinations, the data in Fig. 8 indicate beyond reasonable doubt that the classical 1934 study by Hamer [32] is in error. This is interesting because Hamer’s work was for many years regarded as the most reliable study available of the thermodynamics of sulfate ion protonation; its accuracy was first questioned in 1952 by Davies et al. [33] and subsequently by others. The discrediting of these widely adopted and apparently reliable values serves as a timely reminder of the very real difficulties that bedevil the production of accurate thermodynamic data even in relatively simple systems.
Selection of the most reliable values of \(\Delta_{\text{r}} C_{p,1}^{\text{o}} (T)\) and their uncertainties is difficult because errors increase dramatically with differentiation. A preliminary unweighted assessment of the available data (N = 8) gives \(\Delta_{\text{r}} C_{p,1}^{\text{o}}\)(25 °C) = 257 ± 36 J·K−1·mol−1, with the 1 σ uncertainty corresponding to ± 14%. The data at other temperatures are rather sparse and the uncertainties in \(\Delta_{\text{r}} C_{p,1}^{\text{o}}\) commensurately greater. However, the most important feature of the data in Fig. 8 is the presence of a well-defined minimum in \(\Delta_{\text{r}} C_{p,1}^{\text{o}}\) at T ≈ 100 °C. This confirms the reality of the slight inflections in the \(\log_{10} K_{1}^{\text{o}} (T)\) and \(\Delta_{\text{r}} H_{1}^{\text{o}} (T)\) curves (Figs. 2 and 6). The origin of this minimum is not known but it closely parallels the behavior of the water ionization reaction [23, 24]. It may reflect changes in the water structure and/or the hydration of the ions involved in equilibrium (1). Another possible explanation is pressure change: measurements of \(\Delta_{\text{r}} C_{p,1}^{\text{o}}\) at T ≤ 100 °C are usually made at p = 1 atm, whereas measurements at T > 100 °C must be at p ≥ psat > 1 atm.
3.4 Medium Effects
It is widely appreciated that measurements of thermodynamic data in solutions become increasingly difficult as one moves away from ambient conditions. However, given the large number of publications dealing with the thermodynamics of the first protonation of the sulfate ion, it is surprising that there are insufficient independent experimental data available with which to make a reasonable assessment of the effects of added electrolytes on \(\Delta_{\text{r}} H_{1}^{\text{o}}\), \(\Delta_{\text{r}} S_{1}^{\text{o}}\) or \(\Delta_{\text{r}} C_{p,1}^{\text{o}}\), even under near-ambient conditions. On the other hand, a similar situation appears to pertain to the even more intensively studied water ionization reaction [23].
4 Conclusions
The protonation of the sulfate ion is one of the most-studied equilibria in aqueous solution chemistry. A critical evaluation of the available data has shown that \(K_{1}^{\text{o}}\), the standard state value of the first protonation constant of the sulfate ion, is known to good levels of accuracy up to ~ 250 °C. On the other hand, this equilibrium is less well characterized at higher temperatures and especially in solutions of high ionic strength. Likewise, the enthalpy (ΔrH1), entropy (ΔrS1) and isobaric heat capacity (\(\Delta_{\text{r}} C_{p,1}^{{}}\)) changes accompanying the first sulfate protonation are reasonably well determined at near-ambient temperatures and in dilute solutions. However, these quantities are much more poorly defined at higher temperatures and especially in the presence of even modest concentrations of added electrolytes. Much work remains to be done to characterize the thermodynamic parameters for the first sulfate protonation step under non-standard conditions.
References
Greenwood, N.N., Earnshaw, A.: Chemistry of the Elements, Chap. 15, 2nd edn. Butterworth-Heinemann, Oxford (1997)
Thompson, R.: The Modern Inorganic Chemicals Industry. Chemical Society, London (1977)
Spiro, T.G., Stigliani, W.M.: Chemistry of the Environment, 2nd edn. Prentice Hall, New Jersey (2003)
Burkin, A.R.: Chemical Hydrometallurgy. Imperial College Press, London (2001)
Crompton, T.R.: Battery Reference Book, 3rd edn. Elsevier, Amsterdam (2000)
Jacobs, J.A., Lehr, J.H., Testa, S.M. (eds.): Acid Mine Drainage, Rock Drainage, and Acid Sulfate Soils. Wiley-Blackwell, Hoboken (2014)
Zumdahl, S.S.: Introductory Chemistry: A Foundation, 6th edn. Houghton Mifflin, Boston (2008)
Skoog, D.A., West, D.M., Holler, F.J., Crouch, S.R.: Analytical Chemistry: An Introduction, 7th edn. Saunders College Publishing, Fort Worth (2000)
Noyes, A.A., Eastman, G.W.: In Noyes, A.A.: The Electrical Conductivity of Aqueous Solutions. Carnegie Institution, Washington DC, Publication No. 63, pp. 239–279 (1907)
Staples, B.R.: Activity and osmotic coefficients of aqueous sulfuric acid at 298.15 K. J. Phys. Chem. Ref. Data 10, 779–798 (1981)
Hefter, G.T., Gumiński, C.: Pure Appl. Chem. (in preparation)
Powell, K.J., Brown, P.L., Byrne, R.H., Gajda, T., Hefter, G.T., Leuz, A.-K., Sjöberg, S., Wanner, H.: Sjöberg, S., Wanner, H.: Chemical speciation of environmentally significant metals with inorganic ligands. Part 5: the Zn2+ + OH−, Cl−, \({\text{CO}_{3}^{ 2- }}\), \({\text{SO}_{4}^{ 2- }}\) and \({\text{PO}_{4}^{ 3- }}\) systems. Pure Appl. Chem. 85, 2249–2311 (2013)
Grenthe, I., Puigdomenech, I. (eds.): Modelling in Aqueous Chemistry. OECD, Paris (1997)
Robinson, R.A., Stokes, R.H.: Electrolyte Solutions, 2nd edn. Butterworths, London (1970)
Luther, R.: Die dissociation der schwefelsäuer und arsensäuer (The dissociation of sulfuric acid and arsenic acid). Z. Electrochem. 13, 294–297 (1907)
Kohlrausch, F., Holborn, L.: Das Leitvermögen der Elektrolyte (The Conductivity of Electrolytes), p. 144. Teubner, Leipzig (1898)
Noyes, A.A., Stewart, M.A.: The ionization relations of sulphuric acid. J. Am. Chem. Soc. 32, 1133–1162 (1910)
Clegg, S.L., Brimblecombe, P.: Comment on the “Thermodynamic dissociation constant of the bisulfate ion from Raman and ion interaction modeling studies of aqueous sulfuric acid at low temperatures”. J. Phys. Chem. A 109, 2703–2706 (2005)
Fraenkel, D.: Electrolytic nature of aqueous sulfuric acid. 2. Acidity. J. Phys. Chem. B 116, 11678–11686 (2012)
Sippola, H., Taskinen, P.: Thermodynamic properties of aqueous sulfuric acid. J. Chem. Eng. Data 59, 2389–2407 (2014)
Orwell, G.: Animal Farm. Penguin Books, Middlesex (1965)
Baes, C.F., Mesmer, R.E.: The Hydrolysis of Cations. Wiley, New York (1976)
Brown, P.L., Ekberg, C.: Hydrolysis of Metal Ions. Wiley, Weinheim (2016)
Bandura, A.V., L’vov, S.N.: The ionization constant of water over wide ranges of temperature and density. J. Phys. Chem. Ref. Data 35, 15–30 (2006)
Young, T.F., Irish, D.E.: Solutions of electrolytes. Ann. Rev. Phys. Chem. 13, 435–458 (1962)
Readnour, J.M., Cobble, J.W.: Thermodynamic properties for the dissociation of bisulfate ion and the partial molal heat capacities of bisulfuric acid and sodium bisulfate over an extended temperature range. Inorg. Chem. 8, 2174–2182 (1969)
Dickson, A.G., Wesolowski, D.J., Palmer, D.A., Mesmer, R.E.: Dissociation constant of bisulfate ion in aqueous sodium chloride solutions to 250 °C. J. Phys. Chem. 94, 7978–7985 (1990)
Ashurst, K.G., Hancock, R.D.: Characterization of inner- and outer-sphere complexes by thermodynamics and absorption spectra. Part 1. Sulphato-complexes of the first-row transition elements. J. Chem. Soc. Dalton Trans. 1701–1707 (1977)
Holmes, H.F., Mesmer, R.E.: Isopiestic studies of H2SO4(aq) at elevated temperatures. Thermodynamic properties. J. Chem. Thermodyn. 24, 317–328 (1992)
Chen, H., Irish, D.E.: A Raman spectral study of bisulfate–sulfate systems. II. Constitution, equilibria, and ultrafast proton transfer in sulfuric acid. J. Phys. Chem. 75, 2672–2681 (1971)
Marcus, Y.: Ions in Solution and their Solvation. Wiley, Hoboken (2015)
Hamer, W.J.: The ionization constant and heat of ionization of the bisulfate ion from electromotive force measurements. J. Am. Chem. Soc. 56, 860–964 (1934)
Davies, C.W., Jones, H.W., Monk, C.B.: E.M.F. studies of electrolytic dissociation. Part I. Sulphuric acid in water. Trans. Faraday Soc. 48, 921–928 (1952)
Acknowledgements
This work was funded by the Analytical Division of the International Union of Pure & Applied Chemistry (IUPAC) via the project Stability Constants and Related Thermodynamic Data of Metal-Ion/Sulfate Complexes in Aqueous Solution, and (in part) by Rio Tinto Innovation (Melbourne) and the Australian Government via the Australian Research Council Linkage Grant LP130100991.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hefter, G., Gumiński, C. Remarks on the Evaluation of Thermodynamic Data for Sulfate Ion Protonation. J Solution Chem 48, 1657–1670 (2019). https://doi.org/10.1007/s10953-019-00907-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10953-019-00907-w