
Abstract
The fundamental properties and extraction capability of an ionic liquid (IL), trioctylammonium nitrate ([HTOA][NO3]), for PdII and PtIV, are investigated. At room temperature, [HTOA][NO3] is a solid (melting point: 30.7 °C), but it becomes a liquid (melting point: 16.7 °C) when saturated with water. Water-saturated [HTOA][NO3] exhibits a viscosity of 267.1 mPa·s and an aqueous solubility of 2.821 × 10−4 mol·dm−3 at 25 °C, and can be used as an extraction solvent without dilution. [HTOA][NO3] exhibits an extremely high extraction capability for PdII and PtIV in dilute hydrochloric acid (0.1–2 mol·dm−3 HCl); the distribution ratio reaches 3 × 104 for both the metals. From electrospray ionization mass spectrometry analysis, the species extracted in the IL phase are [PdCl3]− and [PdCl2(NO3)]− for PdII and [PtCl6]2− and [PtCl5]− for PtIV. A majority of the other transition metals are considerably less or marginally extracted into [HTOA][NO3] from a 0.1 mol·dm−3 hydrochloric acid solution. The extraction capacity of [HTOA][NO3] is greater than that of other hydrophobic ILs such as [HTOA]Cl and bis(trifluoromethanesulfonyl)imide-based ILs. The metals extracted into the IL phase are quantitatively back-extracted using an aqueous solution containing thiourea and nitric acid. By controlling the thiourea concentration and shaking time, PdII and PtIV are mutually separated to some extent in the back extraction process. The IL phase used for the back extraction can be reused for the forward extraction of these metals after scrubbing it with an aqueous nitric acid solution.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Platinum-group elements are crucial metals used in industrial processes and products, jewelry, and dental materials [1]. Particularly, palladium, platinum, and rhodium are important as components in catalysts used for automobile exhausts and synthesis of organic chemicals. In addition, palladium and platinum also play a large role in fuel cell technology. Meanwhile, the worldwide demand for these metals is still increasing, which is not satisfied by their supply from natural resources; therefore, there is an increase in the requirement for recycling these metals from waste products [2].
Solvent extraction is one of the important methods to separate, refine, and recycle platinum-group elements in different industries [3]. Dialkyl sulfides and tributyl phosphate are agents typically used for extracting palladium and platinum from hydrochloric acid solutions [3, 4]. These agents are used by dissolving them in organic molecular solvents, leading to health, safety, and environmental problems. On the other hand, ionic liquids (ILs) have attracted attention as one of the alternatives to organic molecular solvents for solvent extraction, because ILs are recognized as green solvents [5].
In addition, ILs are also attractive because some of them exhibit a high extraction capability for ionic species [6,7,8,9,10,11,12,13,14]. The extraction capability of ILs is considerably dependent on the kind of constituent ions, the combinations of which are numerous. To obtain information regarding the rules governing the extraction capability of ILs for ions, the extraction behavior of several ions such as substituted phenolate [14,15,16], tetrachloroaurate [17], and paraquat ions [18] in various IL/aqueous biphasic systems has been investigated. The results revealed that the distribution ratio of each target ion is a function of the aqueous solubility of the IL used; the dependence can be explained on the basis of the extraction mechanism including ion exchange and ion-pair transfer processes with the IL constituent ions. Using an anionic species as the extraction target, for ILs with different anions and a certain cation, the extraction capability increases with an increase in the aqueous solubility of the IL; the opposite holds true for the ILs with different cations and a given anion. Long-chain alkylammonium salts of small inorganic anions such as methyltrioctylammonium chloride ([MTOA]Cl) and trioctylammonium chloride ([HTOA]Cl), which are known as organic anion exchangers, represent typical examples that are thought to exhibit a high extraction capability for an anion [19,20,21]. However, these compounds are solids or highly viscous liquids at room temperature. Hence, they are always used after being diluted with organic molecular solvents for the extraction purpose, although it is unfavorable to use any organic molecular solvent because of safety and environmental concerns. Organic anion exchangers diluted with organic molecular solvents such as toluene and kerosene are known to be effective for extracting platinum-group elements in hydrochloric acid solutions [22,23,24,25].
Neat ILs have been used as extracting phases for platinum group elements in aqueous chloride solutions, in which a majority of the metals exist as anionic chloride complexes [26,27,28,29,30,31,32]. Previously, our group has proposed the use of mixtures with two ILs, that is, trioctylammonium nitrate ([HTOA][NO3]) and trioctylammonium bis(trifluoromethanesulfonyl)imide ([HTOA][NTf2]) [28]. Here [HTOA][NO3], which is expected to be an excellent anion exchanger, is a solid at room temperature; thus, [HTOA][NTf2] is used as a diluent. For example, a mixture of 10 mass% [HTOA][NO3] in [HTOA][NTf2] exhibits a relatively low viscosity (235.7 mPa·s) when saturated with water at 25 °C and has a powerful, selective extraction capability for PdII and PtIV from dilute hydrochloric acid solutions. The use of IL mixtures is expected to exhibit the following merits: ILs exhibit higher hydrophobicity and lower viscosity compared to a pure liquid-ion exchanger; their extraction capability, viscosity, and hydrophobicity can be controlled by the composition ratio; they are nonflammable and environmentally friendly because they only contain ions. In addition, the [HTOA] salts are so-called “protic ILs” that can be easily prepared by simple acid–base reactions of trioctylammonium (TOA) with HNO3 and HNTf2.
If [HTOA][NO3] were a hydrophobic, low-viscosity liquid, it could be used as a powerful extractant without any diluent. In this study, the extraction of PdII, PtIV, and some other heavy metals using water-saturated [HTOA][NO3] from hydrochloric acid solutions was examined because [HTOA][NO3] was found to become a liquid at room temperature when saturated with water, and its viscosity and aqueous solubility were sufficiently low (data are shown below). To the best of our knowledge, this is the first study in which [HTOA][NO3] was used as a metal extractant without dilution. In addition, some other water-saturated ILs were examined for comparison. Fundamental properties such as melting point, density, viscosity, water content, and aqueous solubility of water-saturated [HTOA][NO3] were also investigated.
2 Experimental Section
2.1 Reagents and Instruments
[HTOA][NO3], [HTOA]Cl, [HTOA][NTf2], and 1-butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide ([BMIm][NTf2]) were prepared according to a previously reported study [17, 28]. [MTOA]Cl with a > 97 mass% purity was purchased from Sigma–Aldrich and used as received. Methyltrioctylammonium nitrate ([MTOA][NO3]) was prepared as follows: First, 50 cm3 of a benzene solution containing 1.0 mol·dm−3 of [MTOA]Cl was mixed with 50 cm3 of an aqueous solution containing 4.0 mol·dm−3 of sodium nitrate, and after centrifugation, the benzene phase was separated. This operation was carried out four times for the benzene solution to completely exchange Cl− with \( {\text{NO}}_{3}^{ - } \). Furthermore, the benzene phase was washed five times with deionized water to remove Na+. Then, it was transferred to a boiling flask, evaporated to dryness using a rotary evaporator, and further dried over P2O5 under vacuum for 24 h to afford a pale-yellow solid (16.8 g, 78% yield). 1H NMR (400 MHz, CDCl3, TMS): δ = 3.278–3.365 (t, 6H, CH2), 3.148 (s, 3H, CH3), 1.631–1.751 (m, 6H, CH2), 1.258–1.425 (m, 30H, CH2), 0.850–0.885 (t, 9H, CH3). MS (ESI, acetonitrile, m/z): positive, 368.4240, [MTOA]+; negative, 492.4034, \( \left[ {\text{MTOA}} \right]\left[ {{\text{NO}}_{3} } \right]_{2}^{ - } \). The amounts of Na+ and Cl− in the product were analyzed by atomic absorption spectrophotometry (AAS) and ion-selective potentiometry, respectively (Na+ < 3 ppm, Cl− < 8 ppm). Ultrapure or equivalent grade mineral acids (HCl and HNO3) were purchased from Kanto Chemicals. Standard aqueous solutions of MnII, FeIII, CoII, NiII, CuII, ZnII, RhIII, PdII, and PtIV, purchased as 1000 ppm atomic absorption standards from Kanto Chemicals or Wako Pure Chemicals, were used after appropriate dilution. Dichloromethane and benzene (Kanto Chemicals, guaranteed reagent grade) were purified by distillation and triplicate washing with water, respectively. Sudan III (1-[(E)-{4-[(Z)-phenyldiazenyl]phenyl}diazenyl]-2-naphthol, Tokyo Chemical Industry, > 90 mass% purity), acetonitrile (Kanto Chemicals, LC/MS grade), and other reagents (Kanto Chemicals or Wako Pure Chemicals, guaranteed reagent grade) were used as received. Water was deionized and further purified using a Milli-Q Labo system (Millipore).
A Thermo Neslab RTE-7 thermocontrolled water bath and a Nihon Keiryo Kogyo No.1 primary standard thermometer (uncertainty = ± 0.03 K) were used for melting point measurements. An Anton Paar DMA35n oscillating U-tube density meter, a Kusano No. 2B Ubbelohde-type glass viscometer (viscometer constant: 0.4718 nm2·s−2), and a Hiranuma AQ-7 Karl–Fischer coulometric titrator were used for measurements of density, viscosity, and IL water content, respectively. UV–Vis spectrophotometry and AAS measurements were carried out on a Beckman DU-640 spectrophotometer and a Hitachi Z-6100 polarized Zeeman flame atomic absorption spectrophotometer, respectively. Potentiometric determination of Cl− was carried out on a Horiba F-23 ion meter equipped with a #6560-10C Cl− ion-selective electrode (ISE). The shaking and centrifugation of a centrifuge tube were carried out using a Taitec SR-1N reciprocal shaker (190 strokes·min−1) and a Kubota 2010 tabletop centrifuge (3000 rpm) with an RS-240 swing rotor, respectively. 1H NMR and mass spectral measurements were carried out on a JEOL JNM-ECS400 FT-NMR spectrometer and a Thermo-Fisher Exactive mass spectrometer, respectively, at the Center for Analytical Instrumentation, Chiba University.
2.2 Measurements of Fundamental Properties of Water-Saturated ILs
The melting point of water-saturated [HTOA][NO3] and the density, viscosity, water content, and aqueous solubility of water-saturated [HTOA][NO3], [HTOA]Cl, [MTOA][NO3], and [MTOA]Cl were determined. First, the IL was equilibrated with water in a thermostatic water bath at 25 ± 0.05 °C, and its melting point was measured by the rising melting point method. The density was measured at 25 ± 0.2 °C using a density meter, which was calibrated using pure water. Kinematic viscosity was measured at 25 ± 0.05 °C using a Ubbelohde viscometer calibrated by the manufacturer, and the dynamic viscosity was calculated as the product of the kinematic viscosity and density. The water content was measured by a Karl–Fischer titration, and the concentration of the IL cation in the aqueous phase was measured by the ion-pair extraction and spectrophotometric method using bis[2-(5-bromo-2-pyridylazo)-5-(N-propyl-N-sulfopropylamino)phenolato]cobaltate(III) [33].
The volume change of the IL phase from the equilibration of water-saturated [HTOA][NO3] with hydrochloric acid solutions was evaluated as follows. A solution of 5.0 × 10−4 mol·dm−3 Sudan III dissolved in water-saturated [HTOA][NO3] was prepared. The IL solution of Sudan III was equilibrated with a 100-fold volume of a hydrochloric acid solution (0.10–2.0 mol·dm−3 HCl) in a centrifuge tube by mechanically shaking the tube in a thermostatic chamber at 25 ± 0.2 °C. Following centrifugation for complete phase separation, the tube was allowed to stand for more than 15 min in a thermostatic water bath at 25 ± 0.05 °C. The IL phases before and after shaking were diluted to greater than 20-fold with ethanol, and the Sudan III concentrations were spectrophotometrically determined (λmax = 507.0 nm, ε = (3.15 ± 0.07) × 10−4 cm−1·mol−1·dm3 in ethanol at 25 °C). It was separately confirmed that the partition of Sudan III into the aqueous phase was negligibly small.
In addition, the density of the IL phase, the concentration of Cl− in the IL phase, and the concentrations of [HTOA]+ and [NO3]− in the aqueous phase were determined for the [HTOA][NO3]–hydrochloric acid biphasic system. The density and [HTOA]+ concentration were measured in the same manner as described above. The concentration of \( {\text{NO}}_{3}^{ - } \) in the aqueous phase was determined by the spectrophotometric method using brucine (2,3-dimethoxystrychnidin-10-one) [34]. The concentration of Cl− in the IL phase was determined as follows. A portion of the IL phase was placed in a separate tube and mechanically shaken for 1 h with a 10-fold volume of a 1.0 mol·dm−3 aqueous HNO3 solution; by this operation, Cl− in the IL phase was quantitatively back-extracted into the aqueous HNO3 solution. After centrifugation, a portion of the HNO3 phase was pipetted out and neutralized using an aqueous NaOH solution. The Cl− concentration was measured by an ISE.
2.3 Forward Extraction of Metal Ions
Aqueous solutions containing 0.10–2.0 mol·dm−3 HCl and metal ions were prepared and stored for greater than 24 h at room temperature; the metal concentrations were 1.0 × 10−4 to 7.3 × 10−4 mol·dm−3 for Pd; 9.7 × 10−5 mol·dm−3 for Rh; 7.4 × 10−5 to 5.4 × 10−4 mol·dm−3 for Pt; 1.0 × 10−4 mol·dm−3 for Co and Ni; 9.6 × 10−5 mol·dm−3 for Zn; 9.8 × 10−5 mol·dm−3 for Cu; 1.1 × 10−4 mol·dm−3 for Mn and Fe. A certain amount of a water-saturated IL was weighed in a centrifuge tube; the volume was calculated from the mass using the density (Table 1). A 100-fold volume of the hydrochloric acid solution of a metal ion was added to the centrifuge tube. The tube was mechanically shaken for 15 min to 2 h in a thermostatic chamber at 25 ± 0.2 °C. After centrifugation, the tube was allowed to stand for more than 15 min in a thermostatic water bath at 25 ± 0.05 °C. The metal ion in the aqueous phase was determined by AAS in the flame mode (for Zn) or the graphite furnace mode (for other metals). The extraction percentage (%E) and the distribution ratio (D) were calculated from the following equations:
Here, \( C_{\text{aq}}^{\text{o}} \) and Caq represent the concentrations of metals in the aqueous phase before and after extraction, respectively, and Vaq and VIL are the volumes of the aqueous and IL phases, respectively.
2.4 Mass Spectrometric Analysis of the IL Phase after Extraction
A centrifuge tube, in which a 0.10 mol·dm−3 hydrochloric acid solution containing 1.0 × 10−3 mol·dm−3 of PdII or 1.1 × 10−3 mol·dm−3 of PtIV and water-saturated [MTOA][NO3] were placed in a volume ratio of 100 : 1, was mechanically shaken for 1 h at 25 °C. After centrifugation, a portion of the IL phase was transferred to a sample vial, diluted to ~ 2000-fold with acetonitrile, and subjected to electrospray ionization mass spectrometry (ESI–MS) analysis.
2.5 Back Extraction of Metal Ions
An aliquot of the IL phase after the forward extraction was transferred to another centrifuge tube, to which a 10-fold volume of a stripping solution was added. Here, the stripping solutions examined were as follows: 1.0 mol·dm−3 HCl; 1.0 mol·dm−3 HNO3; 1.0 mol·dm−3 thiourea; 1.0 mol·dm−3 thiourea and 1.0 mol·dm−3 HCl; 0.10–1.0 mol·dm−3 thiourea and 1.0 mol·dm−3 HNO3. The tube was mechanically shaken for 5 min to 4 h at 25 ± 0.2 °C. After centrifugation, the aqueous phase was subjected to AAS. The back extraction percentage (%Eback) was calculated as follows:
where \( C_{\text{B,IL}}^{\text{o}} \) and CB,aq represent the concentrations of metals in the IL phase before back extraction and in the aqueous phase after back extraction, respectively, and VB,aq and VB,IL are the volumes of the aqueous and IL phases used for back extraction, respectively.
3 Results and Discussion
3.1 Fundamental Properties of Water-Saturated ILs
Water saturated [HTOA][NO3] is a liquid at room temperature and its melting point was determined to be 16.7 ± 0.8 °C; this value is lower than that of dry [HTOA][NO3], 30.7 °C [28]. Table 1 summarizes the values of density, viscosity, water content, and aqueous solubility. In addition, the data for other water-saturated ILs, including literature values [28, 35], are summarized. All of these ILs are liquids at 25 °C when saturated with water.
The densities of the Cl−- and \( {\text{NO}}_{3}^{ - } \)-based ILs are less than 1 g·cm−3, that is, around the density of water, whereas those of the [NTf2]−-based ILs are greater than 1 g·cm−3. The viscosity of water-saturated [MTOA][NO3] is comparable to that of [BMIm][PF6] (249.6 mPa·s when dried [36]), showing that water-saturated [MTOA][NO3] can be used as a solvent for extraction without any viscosity issues. All of the other water-saturated ILs, except for [MTOA][NTf2] (485.4 mPa·s [28]), are even less viscous.
For a given cation, the content of water in the IL increases with the anion hydrophilicity as expected from the Gibbs energy for the transfer of the anion from water to nitrobenzene [37]: [NTf2]− < \( {\text{NO}}_{3}^{ - } \) < Cl−. In addition, the aqueous solubility of the IL increases in the same order. For the [NTf2]−-based ILs, the water content increases in the order of [MTOA]+ < [HTOA]+ < [BMIm]+, whereas the aqueous solubility increases in the order of [HTOA]+ < [MTOA]+ < [BMIm]+. These orders mostly correspond to the hydrophilicity order of the cations as expected from the chemical structures, that is, [MTOA]+ < [HTOA]+ < [BMIm]+. However, the water content and aqueous solubility of Cl−- or \( {\text{NO}}_{3}^{ - } \)-based ILs are greater for [MTOA]+ than for [HTOA]+, which is difficult to explain by the hydrophilicity order of the cations. The unexpectedly low water content and aqueous solubility of [HTOA]+-based ILs are possibly related to the strong hydrogen bond interactions of the protic cation with the anions. The aqueous solubility of [HTOA][NO3] is comparable to that of hexane (6.5 × 10−4 mol·dm−3 [38]) and less than that of 1-octanol (4.13 × 10−3 mol·dm−3 [38]) and that of chloroform (6.8 × 10−2 mol·dm−3 [38]). Therefore, water-saturated [HTOA][NO3] is sufficiently hydrophobic to be used as an extraction solvent.
For the [HTOA][NO3]–hydrochloric acid biphasic system (IL/aqueous volume ratio = 1/100), the volume change and density of the IL phase, the concentrations of [HTOA]+ and \( {\text{NO}}_{3}^{ - } \) in the aqueous phase, and the concentration of Cl− in the IL phase were evaluated. This biphasic system is the same as that used for the extraction of metals. Table 2 summarizes the results. With the increase in the initial HCl concentration, the aqueous equilibrium concentration of [HTOA]+ decreases, whereas that of \( {\text{NO}}_{3}^{ - } \) increases; therefore, the molar amount transferred from the IL phase to the aqueous phase is always greater for \( {\text{NO}}_{3}^{ - } \) than for [HTOA]+. On the other hand, at equilibrium, the concentration of Cl− in the IL phase increases with the initial concentration of HCl in the aqueous phase. The above results indicate a partial anion exchange between the IL and aqueous phases. The amount of \( {\text{NO}}_{3}^{ - } \) exchanged with Cl− is estimated to be ~ 30 mol% of the total amount of \( {\text{NO}}_{3}^{ - } \) at an aqueous HCl concentration of 2.0 mol·dm−3. The volume change of the IL phase from water-saturated [HTOA][NO3] to that equilibrated with a 100-fold volume of the hydrochloric acid solution is negligibly small (from −1.8 to 1.4%), related to the extremely small loss of [HTOA]+ into the aqueous phase and the small volume fraction of the anions in the IL phase.
3.2 Forward Extraction of Metal Ions
Forward extraction with water-saturated [HTOA][NO3] was carried out for 1 × 10−4 mol·dm−3 PdII and PtIV in hydrochloric acid solutions (0.10, 1.0, or 2.0 mol·dm−3 HCl) with an IL/aqueous volume ratio of 1/100. It was preliminarily confirmed that the values of D for these metals are not influenced by the standing time of the aqueous phase before extraction (0–240 h) and the time of shaking for extraction (15–120 min). Table 3 summarizes the D and %E values at different HCl concentrations. Except for PtIV at 0.10 mol·dm−3 HCl, these metals are quantitatively extracted (%E ≥ 99%) over all concentrations of HCl; the %E value of PtIV at 0.10 mol·dm−3 HCl is slightly lower (91%). The maximum D values observed are 3.2 × 104 for PdII (at 1.0 mol·dm−3 HCl) and 2.8 × 104 for PtIV (at 2.0 mol·dm−3 HCl). From the maximum D values, the %E values of PdII and PtIV are both estimated to be 97% for a volume ratio of 1/1000 for the IL phase to the aqueous phase, indicating that greater than 1000 fold enrichment of these metals is possible using [HTOA][NO3]. Indeed, the %E value of PdII determined experimentally with an IL/aqueous volume ratio of 1/1000 was 97% from 1.0 mol·dm−3 HCl.
In addition, Table 3 summarizes the D and %E values of other metals. ZnII and FeIII are well extracted at 1.0 and 2.0 mol·dm−3 HCl, but the %E values at 0.1 mol·dm−3 HCl are 50 and 11%, respectively. The %E values of MnII, CoII, NiII, CuII, and RhIII are less than or equal to ~ 10% at 0.1 mol·dm−3 HCl. Therefore, an HCl concentration of 0.1 mol·dm−3 is suitable for the selective extraction of PdII and PtIV.
The extractability order of the metal ions (except for kinetically inert RhIII) corresponds to that of the stability of chloro complexes in the aqueous solution [39]. For example, in a 0.1 mol·dm−3 hydrochloric acid solution, 99% of PdII exists as [PdCl3]− and [PdCl4]2−, whereas almost 100% of NiII exists as the aqua complex, [Ni(H2O)6]2+. To evaluate the extracted species of PdII and PtIV, ESI–MS spectra of the IL phase after extraction were recorded. The spectra obtained in the positive-ion mode exhibited only peaks corresponding to the IL constituent ions. Figures 1 (Pd) and 2 (Pt) show the spectra obtained in the negative-ion mode. In the spectra, there are the peak groups corresponding to [PdCl3]− (calculated monoisotopic m/z = 210.8100) and [PdCl2(NO3)]− (237.8290) for PdII (Fig. 1) and those corresponding to [PtCl6]2− (202.3890), [PtCl5]− (369.8090), and [HTOA][PtCl6]− (759.1879) for PtIV (Fig. 2). In these species, [PdCl3]−, [PdCl2(NO3)]−, and [PtCl5]− are coordinatively unsaturated; therefore, they possibly exhibit weak coordination of a water molecule in the IL phase. Previously [40], the extracted species of PdII and PtIV with chloroform solutions of quaternary ammonium chlorides were thought to be [PdCl3]−, [PdCl4]2−, and [PtCl6]2− from the analysis of the extraction data. The result based on ESI–MS indicate that the extracted species in the present extraction system are more complicated.

Negative-ion ESI–MS spectra of the IL phase (diluted with acetonitrile) after the extraction of 1.0 × 10−3 mol·dm−3 PdII in 0.10 mol·dm−3 hydrochloric acid with a 1/100 volume of [HTOA][NO3]. Measuring range: m/z = 50–800

Negative-ion ESI–MS spectra of the IL phase (diluted with acetonitrile) after the extraction of 1.1 × 10−3 mol·dm−3 PtIV in 0.10 mol·dm−3 hydrochloric acid with a 1/100 volume of [HTOA][NO3]. Measuring range: m/z = 50–800
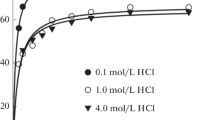
The extractability of PdII and PtIV from 0.1 mol·dm−3 hydrochloric acid solutions to water-saturated [HTOA][NO3] was investigated with different metal ion concentrations. Figure 3 shows the D values as a function of the initial metal concentration in the aqueous phase. There is no significant dependence of D on the metal concentration. This result is consistent with the fact that the extracted species are all mononuclear complexes.

Distribution ratios of PdII (filled circle) and PtIV (open triangle) as a function of the initial metal concentration in the aqueous phase (0.10 mol·dm−3 hydrochloric acid). IL/aqueous volume ratio: 1/100. Shaking time: 1 h
To compare the extraction capabilities of different ILs, the extractability of PdII and PtIV from 0.1 mol·dm−3 hydrochloric acid solutions to several water-saturated ILs was investigated. Table 4 summarizes the D and %E values, where the data for [MTOA]Cl are absent because the aqueous phase changed to cloudy due to the formation of micro-emulsions. The extractability is clearly considerably dependent on the IL: extractability increases in the order of [BMIm][NTf2] < [MTOA][NTf2] < [HTOA][NTf2] < [HTOA]Cl < [MTOA][NO3] < [HTOA][NO3] for PdII and [BMIm][NTf2] < [MTOA][NTf2] < [HTOA][NTf2] < [HTOA]Cl < [HTOA][NO3] < [MTOA][NO3] for PtIV. The extraction capability of [HTOA][NO3] is the highest for PdII and the second highest for PtIV. As described in the Introduction, for the ILs composed of different anions and a specific cation, the extraction capability for a target anion increases with the increase in the IL aqueous solubility; the opposite holds true for the ILs composed of different cations and a specific anion [14,15,16,17]. The above orders of the ILs are almost consistent with those expected from the aqueous solubility values of the ILs (Table 1). This fact also supported that PdII and PtIV are extracted as anionic species. The higher extraction capability of [HTOA][NO3] compared with that of [HTOA]Cl is not expected. Particularly, for the extraction of PdII, the D value with [HTOA][NO3] is more than 20-fold greater than that with [HTOA]Cl, possibly related to the extraction of [PdCl2(NO3)]− as suggested by the ESI–MS analysis. In addition, notably, PtIV is extracted to a greater extent compared to PdII using [BMIm][NTf2] and [MTOA][NTf2], whereas PdII is extracted with the other ILs to a greater extent although the reason is not clear at this stage.
3.3 Back Extraction of Pd and Pt
Some aqueous stripping solutions were examined for the back extraction of PdII and PtIV from the [HTOA][NO3] phase after forward extraction. The IL/aqueous volume ratio and shaking time for back extraction were kept constant at 1/10 and 1 h, respectively. Table 5 summarizes the %Eback values. PdII and PtIV are not back-extracted or marginally back-extracted with a 1.0 mol·dm−3 HCl or HNO3 aqueous solution, respectively. On the other hand, a 1.0 mol·dm−3 aqueous thiourea solution exhibits an extremely good back extraction efficiency for these metals. An aqueous solution containing both 1.0 mol·dm−3 of thiourea and 1.0 mol·dm−3 of HNO3 exhibits a higher back extraction efficiency, probably related to anion exchange, that is, the exchange of the chloro-complex anions of the metals in the IL phase with \( {\text{NO}}_{3}^{ - } \) in the aqueous phase. However, an aqueous solution containing both 1.0 mol·dm−3 of thiourea and 1.0 mol·dm−3 of HCl exhibits a lower back extraction efficiency compared with that of an aqueous solution containing only 1.0 mol·dm−3 of thiourea, because Cl− contributes to the forward extraction of these metals. In addition, back extraction experiments of PdII and PtIV were conducted using 1.0 mol·dm−3 aqueous HNO3 solutions containing different concentrations of thiourea (0.10 and 0.50 mol·dm−3). Basically, the %Eback values of the metals decrease with a decrease in the thiourea concentration, but PdII is quantitatively back-extracted even at a thiourea concentration of 0.50 mol·dm−3. The %Eback value is always greater for PdII than for PtIV.
The effect of the shaking time for back extraction with an aqueous solution containing 0.50 mol·dm−3 of thiourea and 1.0 mol·dm−3 of HNO3 was evaluated at an IL/aqueous volume ratio of 1/10. Figure 4a shows the plot of %Eback versus shaking time. PdII is quantitatively back-extracted with shaking time ≥ 30 min. In addition, the quantitative back extraction of PtIV is achieved by shaking for 2 h. The back extraction rate of PtIV is clearly slower than that of PdII. Then, the effect of the shaking time for back extraction was investigated again using an aqueous HNO3 solution (1.0 mol·dm−3) containing more dilute thiourea (0.10 mol·dm−3), expecting the separation of PdII from PtIV. Figure 4b shows the result. The %Eback values are 86% for PdII and 23% for PtIV by shaking for 15 min and 91% for PdII and 34% for PtIV by shaking for 30 min. The mutual separation between PdII and PtIV by back extraction is not complete but possible to some extent.

Effect of shaking time on the back extraction percentages of PdII (filled circle) and PtIV (open triangle) from the IL phase ([HTOA][NO3]) using a 1.0 mol·dm−3 aqueous HNO3 solution containing thiourea. Initial metal concentration in the IL phase: 9.9 × 10−3 mol·dm−3 (Pd), 1.0 × 10−2 mol·dm−3 (Pt). IL/aqueous volume ratio: 1/10. Thiourea concentration: (a) 0.50 mol·dm−3 and (b) 0.10 mol·dm−3
3.4 Regeneration of [HTOA][NO3]
During forward extraction, NO3− in the IL phase is partially exchanged with Cl− as shown in Table 2. However, during back extraction using an aqueous thiourea solution containing HNO3, Cl− in the IL phase should be reexchanged with \( {\text{NO}}_{3}^{ - } \). Therefore, [HTOA][NO3] is expected to be regenerated by back extraction and reusable for the forward extraction of PdII and PtIV. To verify this, the following experiments were conducted. Assuming forward and back extraction, water-saturated [HTOA][NO3] was shaken for 1 h at 25 °C with a 100-fold volume of 0.10 mol·dm−3 hydrochloric acid (not containing metals) and, after centrifugation, an aliquot of the IL phase was shaken for 2 h at 25 °C with a 10-fold volume of an aqueous solution containing 0.50 mol·dm−3 of thiourea and 1.0 mol·dm−3 of HNO3. After centrifugation, the IL phase was used for the extraction of PdII or PtIV in a 100-fold volume of 0.10 mol·dm−3 hydrochloric acid. However, both metals were not well extracted, probably because of the presence of thiourea in the IL phase. Then, to remove thiourea, the IL phase was shaken with a 10-fold volume of an aqueous solution of 1.0 mol·dm−3 HNO3 for 1 h. Using the IL phase after the scrubbing, 99% of PdII and 88% of PtIV were extracted during forward extraction from a 100-fold volume of a 0.10 mol·dm−3 hydrochloric acid solution. These %E values are nearly equal to those obtained using fresh water-saturated [HTOA][NO3] (Table 3), indicating the successful regeneration of [HTOA][NO3].
4 Conclusion
At room temperature, water-saturated [HTOA][NO3] is a liquid with a relatively low viscosity and sufficiently low aqueous solubility, which can be used as an extraction solvent without dilution. It has an extremely high extraction capability for PdII and PtIV in dilute hydrochloric acid, which is sufficient to quantitatively extract these metals from a 1000-fold volume of the aqueous phase. Other transition metals (MnII, FeIII, CoII, NiII, CuII, ZnII, and RhIII) are considerably less or marginally extracted into [HTOA][NO3] from the dilute hydrochloric acid solution. The extraction capability of [HTOA][NO3] is generally greater than those of the other hydrophobic ILs examined, such as [HTOA]Cl and several [NTf2]−-based ILs. The metals extracted in the [HTOA][NO3] phase are quantitatively back-extracted using an aqueous solution containing thiourea and HNO3. By controlling the thiourea concentration and shaking time, the mutual separation of PdII and PtIV is also possible to some extent during back extraction. [HTOA][NO3] can be readily, cost-effectively prepared by the neutralization of trioctylamine with nitric acid and can be regenerated via scrubbing the used IL with an aqueous HNO3 solution. Consequently, this extraction system can be possibly applied for the industrial separation processes and preconcentration prior to the instrumental analysis of PdII and PtIV.
References
PGM Market Report May 2017. Johnson Matthey Precious Metals Management, Royston (2017)
Fornalczyk, A., Saternus, M.: Removal of platinum group metals from the used auto catalytic converters. Metalurgija 48, 133–136 (2009)
Bernardis, F.L., Grant, R.A., Sherrington, D.C.: A review of methods of separation of the platinum-group metals through their chloro-complexes. React. Funct. Polym. 65, 205–217 (2005)
Cox, M.: Solvent extraction in hydrometallurgy. In: Rydberg, J., Cox, M., Musikas, C., Choppin, G.R. (eds.) Solvent Extraction Principles and Practice, 2nd edn, pp. 454–503. CRC/Taylor and Francis, Boca Raton (2004)
De Los Rios, A.P., Hernandez-Fernandez, F.J. (eds.): Ionic Liquids in Separation Technology. Elsevier, Amsterdam (2014)
Dai, S., Ju, Y.H., Barnes, C.E.: Solvent extraction of strontium nitrate by a crown ether using room-temperature ionic liquids. J. Chem. Soc. Dalton Trans. 1999, 1201–1202 (1999)
Visser, A.E., Swatloski, R.P., Griffin, S.T., Hartman, D.H., Rogers, R.D.: Liquid/liquid extraction of metal ions in room temperature ionic liquids. Sep. Sci. Technol. 36, 785–804 (2001)
Carda-Broch, S., Berthod, A., Armstrong, D.W.: Solvent properties of the 1-butyl-3-methylimidazolium hexafluorophosphate ionic liquid. Anal. Bioanal. Chem. 375, 191–199 (2003)
Vidal, S.T.M., Correia, M.J.N., Marques, M.M., Ismael, M.R., Reis, M.T.A.: Studies on the use of ionic liquids as potential extractants of phenolic compounds and metal ions. Sep. Sci. Technol. 39, 2155–2169 (2005)
Khachatryan, K.S., Smirnova, S.V., Torocheshnikova, I.I., Shvedene, N.V., Formanovsky, A.A., Pletnev, I.V.: Solvent extraction and extraction-voltammetric determination of phenols using room temperature ionic liquid. Anal. Bioanal. Chem. 381, 464–470 (2005)
Vijayaraghavan, R., Vedaraman, N., Surianarayanan, M., MacFarlane, D.R.: Extraction and recovery of azo dyes into an ionic liquid. Talanta 69, 1059–1062 (2006)
Pei, Y.C., Wang, J.J., Xuan, X.P., Fan, J., Fan, M.: Factors affecting ionic liquids based removal of anionic dyes from water. Environ. Sci. Technol. 41, 5090–5095 (2007)
de los Rios, A.P., Hernandez-Fernandez, F.J., Lozano, L.J., Sanchez, S., Moreno, J.I., Godinez, C.: Removal of metal ions from aqueous solutions by extraction with ionic liquids. J. Chem. Eng. Data 55, 605–608 (2010)
Katsuta, S., Nakamura, K., Kudo, Y., Takeda, Y., Kato, H.: Partition behavior of chlorophenols and nitrophenols between hydrophobic ionic liquids and water. J. Chem. Eng. Data 56, 4083–4089 (2011)
Katsuta, S., Nakamura, K., Kudo, Y., Takeda, Y.: Mechanisms and rules of anion partition into ionic liquids: phenolate ions in ionic liquid/water biphasic systems. J. Phys. Chem. B 116, 852–859 (2012)
Watanabe, Y., Katsuta, S.: Distribution of a monovalent anion in various ionic liquid/water biphasic systems: relationship of the distribution ratio of picrate ions with the aqueous solubility of ionic liquids. J. Chem. Eng. Data 59, 696–701 (2014)
Katsuta, S., Watanabe, Y., Araki, Y., Kudo, Y.: Extraction of gold(III) from hydrochloric acid into various ionic liquids: relationship between extraction efficiency and aqueous solubility of ionic liquids. ACS Sustain. Chem. Eng. 4, 564–571 (2016)
Hamamoto, T., Okai, M., Katsuta, S.: The laws governing ionic liquid extraction of cations: partition of 1-ethylpyridinium monocation and paraquat dication in ionic liquid/water biphasic systems. J. Phys. Chem. B 119, 6317–6325 (2015)
Seeley, F.G., Crouse, D.J.: Extraction of metals from chloride solutions with amines. J. Chem. Eng. Data 11, 424–429 (1966)
Galan, B., Urtiaga, A.M., Alonso, A.I., Irabien, J.A., Ortiz, M.I.: Extraction of anions with Aliquat 336: chemical equilibrium modeling. Ind. Eng. Chem. Res. 33, 1765–1770 (1994)
Lee, M.-S., Lee, K.-J., Oh, Y.-J.: Solvent extraction equilibria of FeCl3 from hydrochloric acid solution. Mater. Trans. 45, 2364–2368 (2004)
Jha, M.K., Gupta, D., Lee, J.-C., Kumar, V., Jeong, J.: Solvent extraction of platinum using amine based extractants in different solutions: a review. Hydrometallurgy 142, 60–69 (2014)
Cieszynska, A., Wisniewski, M.: Extraction of palladium(II) from chloride solutions with Cyphos® IL 101/toluene mixtures as novel extractant. Sep. Purif. Technol. 73, 202–207 (2010)
Wei, W., Cho, C.-W., Kim, S., Song, M.-H., Bediako, J.K., Yun, Y.-S.: Selective recovery of Au(III), Pt(IV), and Pd(II) from aqueous solutions by liquid–liquid extraction using ionic liquid Aliquat-336. J. Mol. Liq. 216, 18–24 (2016)
Fontàs, C., Salvadó, V., Hidalgo, M.: Solvent extraction of Pt(IV) by Aliquat 336 and its application to a solid supported liquid membrane system. Solvent Extr. Ion Exch. 17, 149–162 (1999)
Papaiconomou, N., Lee, J.-M., Salminen, J., von Stosch, M., Prausnitz, J.M.: Selective extraction of copper, mercury, silver, and palladium ions from water using hydrophobic ionic liquids. Ind. Eng. Chem. Res. 47, 5080–5086 (2008)
Stojanovic, A., Kogelnig, D., Fischer, L., Hann, S., Galanski, M., Groessl, M., Krachler, R., Keppler, B.K.: Phosphonium and ammonium ionic liquids with aromatic anions: synthesis, properties, and platinum extraction. Aust. J. Chem. 63, 511–524 (2010)
Katsuta, S., Yoshimoto, Y., Okai, M., Takeda, Y., Bessho, K.: Selective extraction of palladium and platinum from hydrochloric acid solutions by trioctylammonium-based mixed ionic liquids. Ind. Eng. Chem. Res. 50, 12735–12740 (2011)
Génand-Pinaz, S., Papaiconomou, N., Leveque, J.-M.: Removal of platinum from water by precipitation or liquid–liquid extraction and separation from gold using ionic liquids. Green Chem. 15, 2493–2501 (2013)
Yang, Y., Kubota, F., Baba, Y., Kamiya, N., Goto, M.: One step effective separation of platinum and palladium in an acidic chloride solution by using undiluted ionic liquids. Solvent Extr. Res. Dev. Jpn. 21, 129–135 (2014)
Tong, Y., Wang, C., Huang, Y., Yang, Y.: Extraction and stripping of platinum from hydrochloric acid medium by mixed imidazolium ionic liquids. Ind. Eng. Chem. Res. 54, 705–711 (2015)
Papaiconomou, N., Svecova, L., Bonnaud, C., Cathelin, L., Billard, I., Chainet, E.: Possibilities and limitations in separating Pt(IV) from Pd(II) combining imidazolium and phosphonium ionic liquids. Dalton Trans. 44, 20131–20138 (2015)
Kasahara, I., Kanai, M., Taniguchi, M., Kakeba, A., Hata, N., Taguchi, S., Goto, K.: Bis[2-(5-bromo-2-pyridylazo)-5-(N-propyl-N-sulphopropylamino)phenolato]cobaltate(III) as a counter ion for the extraction and spectrophotometric determination of long-chain quaternary ammonium salts and tertiary alkylamines in the presence of each other. Anal. Chim. Acta 219, 239–245 (1989)
Saito, G., Sugimoto, K., Hagino, K.: Spectrophotometric determination of nitrate and/or nitrite using brucine sulfate. Bunseki Kagaku 20, 542–549 (1971)
Nakamura, K., Kudo, Y., Takeda, Y., Katsuta, S.: Partition of substituted benzenes between hydrophobic ionic liquids and water: evaluation of interactions between substituents and ionic liquids. J. Chem. Eng. Data 56, 2160–2167 (2011)
Tokuda, H., Tsuzuki, S., Susan, M.A.B.H., Hayamizu, K., Watanabe, M.: How ionic are room-temperature ionic liquids? An indicator of the physicochemical properties. J. Phys. Chem. B 110, 19593–19600 (2006)
Kakiuchi, T., Nishi, N.: Ionic liquid|water interface: a new electrified system for electrochemistry. Electrochemistry 74, 942–948 (2006)
Riddick, J.A., Bunger, W.B., Sakano, T.K.: Organic Solvents, 4th edn. Wiley, New York (1986)
Högfeldt, E. (ed.): Stability Constants of Metal-Ion Complexes Part A: Inorganic Ligands. Pergamon Press, Oxford (1982)
Kondo, K., Ourachi, T., Kaneiwa, T., Matsumoto, M.: Solvent extraction of precious metals with quaternary ammonium salts and its application to preparation of metal particles. Solvent Extr. Res. Dev. Jpn. 7, 176–184 (2000)
Acknowledgements
The authors thank Ms. Sayaka Kado (Center for Analytical Instrumentation, Chiba University) for her technical support on mass spectrometry. The authors also thank Enago (www.enago.jp) for the English language review. This work was financially supported by JSPS KAKENHI Grant Number JP26410145.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Katsuta, S., Tamura, J. Extraction of Palladium(II) and Platinum(IV) from Hydrochloric Acid Solutions with Trioctylammonium Nitrate Ionic Liquid without Dilution. J Solution Chem 47, 1293–1308 (2018). https://doi.org/10.1007/s10953-018-0745-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10953-018-0745-9