
Abstract
The hydrothermal reduction of nitrogen is important in several fields. Hydrazine and amines are injected in the secondary circuit of the pressurized water reactors (PWR) nuclear plants to minimize materials corrosion. Amines are also possible reactants in the nitrogen chemistry of submarine hydrothermal vents. To better evaluate the chemical properties of N-bearing hydrothermal solutions, experiments have been carried out at 275 °C using a hydrothermal reactor equipped for a potentiometric monitoring, allowing in situ pH and Eh measurements. The thermal decomposition of hydrazine in simulated feed waters steam generators of PWR plants was monitored and appeared to be catalyzed by the surface of the titanium vessel or by magnetite particles, present as a model of the usual corrosion products. Rates are also dependent on pH or, keeping in mind that it is a catalyzed reaction, on the increasing proportion of N2H4 relative to N2H +5 , the hydrazinium cation being less sorbed under acidic conditions. Ethanolamine appears to be stable for over a month while morpholine is decomposed in few days in contact with magnetite. The in situ pH measurements are consistent with theoretical predictions using acidity constants of morpholine and ethanolamine from the limited literature data. The redox potential imposed by hydrazine was measured and was between −0.45 V (pHT = 5) and −0.65 V (pHT = 7) provided that the concentration of hydrazine exceeds the threshold concentration of 20–50 μg·kg−1. Above this concentration, the potential was poorly dependent on the hydrazine concentration. The observed pH–Eh relationship reflects proton production during the oxidation of N2H4 to N2. From the Eh values, a standard potential of −1.1 V at 275 °C has been calculated for this reaction.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The aqueous chemistry of nitrogen in hydrothermal conditions, in particular its reduction, is a major concern in several fields, from its use as corrosion inhibitor in the nuclear industry to investigations on the emergence of life. Abiotic nitrogen reduction on the early earth is considered as a constraint for the emergence of life [1]. But, the complex chemistry operating in the hydrothermal vents of the ocean ridge has also drawn the attention of the scientific community, for several decades, due to its potential in prebiotic chemistry [2–4]. Although amino acids are considered to be unstable at high temperature, they are found in hydrothermal vents and their presence is mostly attributed to microbiological activity (e.g., [5]). However, the question of prebiotic chemistry in the vicinity of black smoker type hydrothermal vents is still an open question. For example, Stephen-Sherwood et al. [6] used hydrazine, uracil and formaldehyde as reactants for the possible prebiotic synthesis of thymine. In this complex chemistry, strong reducers like hydrazine are of particular interest but its behavior at high temperature is poorly documented. Additionally, the use of hydrazine as corrosion inhibitor in nuclear power plants is a motivation for fundamental research in this field.
Nuclear power plants use a complex technology, based on the heat transfer from the reactor core to a steam driven alternator. In pressurized water reactors (PWR), steam is generated in a closed circuit, named the secondary circuit. The temperature varies typically from 30 °C in the colder zone (monophasic, aqueous) to 275 °C in the steam generator (biphasic aqueous–vapor). Except in the steam generator itself, the fluid is a diluted aqueous phase under high velocity fluid dynamics. In these conditions, flow accelerated corrosion occurs on carbon steel pipelines of the feed water system and produces corrosion products, essentially magnetite, which may provoke to the fouling and blockage of the steam generator. Hydrazine (N2H4) is injected in the aqueous phase of the secondary circuit in order to minimize materials corrosion, by eliminating oxygen and maintaining a reducing potential. Since hydrazine is not stable at the operational temperature of the steam generators, it must be injected continuously. A complete knowledge of the chemical properties of hydrazine, and companion amines such morpholine and ethanolamine used as pH buffer, is essential for the optimization of the industrial process.
At room temperature, the acid–base and redox properties of hydrazine are well known. In aqueous medium, two successive protonation reaction of hydrazine can take place, with acidity constants 7.2 and −0.4 for pK a2 and pK a1, respectively [7]. Thus, in the cold part of the secondary circuit conditions of PWR (pH25 °C between 9.2 and 9.6), the dominant form is N2H4, along with a small fraction of N2H +5 . The dissociation constant of hydrazinium decreases with the increase of temperature [8], more strongly than the dissociation constant of water, in a way such that N2H +5 doesn’t exist in near neutral and alkaline media above 200 °C. Under the conditions of the hot part of the secondary circuit (275 °C), pH is buffered by the morpholine–ammonia or ethanolamine–ammonia mixtures. The resulting pH may be estimated by using the dissociation constant of ammonium [9] and the acidity constants of morpholine [10] and ethanolamine [11]. The typical in situ values of pH expected in operational conditions of steam generators are between 6.0 and 6.5.
The redox properties of hydrazine are more complex than the acid–base ones. The redox reaction is given by one of the following equations, the oxidation reaction being favored by an alkaline medium:
Lee [12] calculated standard potentials of oxidation–reduction of the N2/N2H4 and N2H4/NH3 systems up to 288 °C. Moreover, hydrazine can undergo a disproportionation [13]. As a consequence of these various possible reactions, the potential imposed by hydrazine at high temperature is difficult to estimate. Only one corrosion potential measurement, on tubes made of alloy 800 in a steam generator of PWR, versus hydrazine concentration in feed water at 270 °C, pHT = 6.5, is given by Stellwag and Killian [14] and indicates a potential around −0.5 V for hydrazine concentrations higher than 50 μg·kg−1.
The main drawback of hydrazine is its rapid thermal decomposition. The byproducts of this reaction (ammonia, nitrogen, hydrogen) modify the pH and the electrochemical potential of the solution. Several reactions describe hydrazine decomposition but two main reactions are observed [15, 16]:
Tests carried out in nuclear power plants showed that hydrazine decomposition produces hydrogen (reaction 4) in the presence of copper. Without copper alloy, reaction (3) occurs. Several reaction paths exist for hydrazine decomposition and they depend on several parameters (type of alloy present, pH, temperature…). In all cases, literature data agree with the fact that the kinetics of hydrazine decomposition is first order. Among all of the pertinent parameters, temperature is the most striking. Thus, the thermal decomposition begins above 160° and increases significantly above 200 °C [17]. As an example, the hydrazine decomposition rate varies by two orders of magnitude for a temperature increase from 270 to 298 °C [18]. However, an accurate determination of the decomposition rate is difficult to determine because this reaction is generally catalyzed (heterogeneous catalysis) by the metallic surfaces in contact. Without metals or oxides to catalyze the reaction, the decomposition rate is too low to be measurable below 270 °C [17]. In the presence of catalytic surfaces (alloys, metal oxides), the hydrazine decomposition rate is also dependent on the surface characteristic and the hydrodynamics of the system. The third parameter is pH. The speciation of hydrazine in aqueous solutions is expected to influence its decomposition rate although this factor is poorly documented, especially at high temperature. It has been reported that a significant decrease of the decomposition rate occurs at 25 °C when the pH decreases from 12 to 8 [17]. This influence of pH seems also to be confirmed at high temperature by a single study conducted by Delaunay et al. [19].
The conclusions of this short review underline the complexity of the chemical reactions involving hydrazine in aqueous solution, and even the absence of direct measurements of fundamental properties of the conditioning media of the secondary circuit such as pH and the redox potential (Eh). This lack of direct measurements is the consequence of the instrumentation limitations. However, the evaluation of pH and Eh at high temperature, after sampling and cooling of the solution, may be affected by the interaction with the atmosphere when measured at room temperature. In situ measurements with glass electrodes are restricted to temperatures lower than 130 °C for obvious mechanical reasons. High temperature pH measurement was early developed in the 1970s by Mesmer et al. [20] with further improvements [21, 22]. This technology uses a hydrogen electrode concentration cell and needs to add a given pressure of hydrogen gas in the reactor. This technology is inadequate for systems sensitive to redox reaction such as a simulated secondary circuit of PWRs containing amines and oxide particles in suspension. With the recent development of oxygen-ion conducting ceramics sensor coupled with an Ag/AgCl reference electrode [23–25], new perspectives are offered for the determination of fluid chemistry at high temperature [26].
This paper presents an experimental investigation of the chemical properties of a simulated secondary circuit of PWR, including acid–base and redox properties, as well as the decomposition rate of hydrazine as a function of pH and catalyst surfaces. The measurements were performed at 275 °C, in the presence or absence of suspended magnetite particles, in a batch or mixed flow titanium reactor. The hydrothermal device was equipped with a Pt redox sensor, a ZrO2 ceramic based pH sensor and an Ag/AgCl reference electrode. The aqueous solution was a mixture of morpholine, ethanolamine, ammonia, hydrazine and potassium chloride as background electrolyte.
2 Materials and Methods
2.1 Description of the Reactor
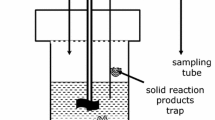
We developed a specific experimental mixed flow system allowing a continuous renewal of the solution and the monitoring of in situ pH and Eh. The experimental design, presented in Fig. 1, is based on a commercial reactor (Parr Instrument®) made up of pure titanium (grade 4). The nominal maximum pressure and temperature of the reactor is 10 MPa and 300 °C. The internal volume of the vessel is 450 mL and it is stirred by an impeller, driven by an external magnetic driver. The temperature is controlled with an immersed thermocouple and is held constant to within 1 °C by an electric furnace. The vessel is equipped with several connections that enable injection of fluid or periodic on-line sampling of the experimental fluids. Mixed flow mode is obtained by injecting the solution at the bottom of the cell with a HPLC pump and continuously withdrawing the excess solution at the top of the cell, the pressure being maintained at a steady-state level by the back-pressure regulator. Mixed-flow experiments were conducted above the vapor pressure, at 10 MPa, so in a single liquid phase. Aliquots of the effluent solution were sampled at the outlet of the back-pressure regulator. Batch experiments were carried out under the vapor pressure.

Experimental setup
2.2 Description of the Electrodes
The continuous in situ pH and Eh monitoring was performed with commercial probes designed and distributed by Corr Instruments®, USA. The potentiometric system consisted in three probes (pH, Eh and reference) with all tubing and connectors being made of pure titanium. The probes were inserted vertically at the top of the measurement cell as shown in Fig. 1. The technical specifications of the probes are the following:
-
The redox probe constituted of a platinum rod in contact with the liquid phase is placed inside the measurement cell and is isolated from the titanium body by a polytetrafluoroethylene (PTFE) line.
-
The ZrO2-based pH probe composed of a Y-doped zirconia tubing containing a dry Ni/NiO internal reference. The pH of the experimental solution influences the electrical reactivity of the zirconia tube, which formally acts as an Ni/NiO/ZrO2(Y2O3)/H+ sensor. This method was originally described by Niedrach [23], and was more recently reviewed in [25].
-
The reference electrode consisted in a PTFE tube filled with a 0.1 mol·kg−1 KCl solution and containing an Ag/AgCl couple in the colder zone at the top of the electrode. The temperature of this zone was maintained at 25 °C by circulating an external cooling fluid. Ionic exchanges between the reference media and the aqueous phase inside the measurement cell were allowed through a ZrO2 frit.
The thermal gradient along the probe is particularly important for the reference electrode filled with an aqueous solution, since a Soret effect (thermophoresis) can lead to a shift in the raw potential of the reference electrode. For pH, the use of a calibration curve with solutions of well known in situ pH made any correction unnecessary. For Eh, corrections were required and are described below.
2.3 Electrodes Calibration
The response of the pH electrode was checked at 225, 250 and 275 °C by using HCl and NaOH solutions of known pH. Several aliquots of the sampled solutions were withdrawn during the course of the experiments and quenched to room temperature. The in situ pH values were calculated from those measured at 25 °C using JChess software [27] and its associated database (see modeling section). The relationship between the in situ pH and the measured potential of the pH probe, relative to the reference probe, is shown in Fig. 2. The effect of the temperature on the pH–mV relationship is weak and the slope of the calibration curve is consistent with Nernstian behavior. The calibration procedure was repeated at regular time interval to ensure the absence of any deviation with time.

Example of pH calibration at 220, 250 and 275 °C. The potential is measured between the pH sensor at the temperature of the process and the Ag/AgCl couple maintained at room temperature
The performance of the redox probe was regularly checked at room temperature with conventional redox buffers, but a calibration procedure at high temperature was not possible in the absence of available buffers for the 250–275 °C temperature range. However, given the simplicity of the redox probe design (a single Pt rod), the temperature effect on the redox measurements is assumed to be related only to the reference probe, through the conversion from Ag/AgCl to normal hydrogen electrode (NHE) potential, liquid junction potential and Soret effect. The Soret effect is induced by the thermal gradient between the process temperature (275 °C for this study) and the temperature of the Ag/AgCl couple located outside of the reactor. To ensure a steady state temperature gradient, and so a steady state Soret effect, the top of the reference probe containing the Ag/AgCl couple was maintained at room temperature using an external cooler. Two previous experimental studies using an external pressure balanced reference electrode have proposed a correction function to convert the measured potential of the cold Ag/AgCl reference probe to the potential of a NHE electrode at the process temperature. Table 1 reports the correction values at different temperatures according to MacDonnald et al. [28] and Bosch et al. [29]. The two correction functions differ by approximately 30 mV. In this work we have followed the most recent study [29] and have applied a systematic correction of −2 mV at 275 °C.
2.4 Fluid Chemistry
The chemistry of the secondary circuit of PWR plants has been reproduced for two typical alkaline conditionings with morpholine or ethanolamine as pH buffer. The composition is detailed in Table 2. KCl and KOH, not used in PWRs, were added in our study to ensure a constant ionic strength and to compensate for the sorption of OH− ions by the reactor walls. Some runs were conducted with KOH in both starting and feed solutions, others with KOH in the starting solution only, still others without KOH. The solutions were prepared from pure chemical reagents in an anoxic glove box.
2.5 Solids
Magnetite was used in few runs at a concentration of 1 g·L−1. The magnetite powder used in this study is an aliquot of the Alfa Aesar sample purified and characterized as previously detailed [30, 31]. Briefly, it consists in agglomerates of facetted particles with a diameters between 100 and 500 nm and its specific surface area, calculated from adsorption of krypton, is 1.68 ± 0.02 m2·g−1.
2.6 Chemicals
The water used in the experiments was deionized water generated by a Direct-Q3 generator from Millipore©, with a resistivity of 18.2 mΩ, and is indicated by “Milli-Q water”. The gradient pure chemicals were supplied by Merck© with grade “Suprapur” for KCl and KOH salts and ammonia solution, and with grade “For Synthesis” for pure morpholine and ethanolamine. Argon gas of grade “Alphagaz1” (C n H m < 0.5 ppm, O2 < 2 ppm), supplied by Air Liquide©, was used for degassing the solutions.
2.7 Experimental
Two kinds of experiments were conducted: (1) monitoring of the decomposition of N2H4 and amines with time in a closed system at 275 °C, and (2) measurement of pH and Eh with a steady state concentration of N2H4 in mixed-flow mode.
The first set of experiments, conducted in a closed system, was the easier to implement. Prior to the transfer to the reactor, 1–10 mg·kg−1 of hydrazine (N2H4) was added to the Milli-Q water in a glove box ([O2] < 10 mL·m−3). The reactor was flushed with Ar gas before heating to eliminate the atmospheric oxygen. The thermal decomposition of N2H4 was monitored by the sampling and the analysis for hydrazine concentration of aliquots of the solution at various elapsed times (see analytical section below). Supplementary runs were conducted at more alkaline pHs or in the presence of iron oxide, by adding magnetite powder. The stability of morpholine and ethanolamine was checked in dedicated KCl-free experiments.
The second set of experiments was conducted with a continuous renewal of the solution to compensate the thermal decomposition of hydrazine. The input solution, as well as the HPLC pump, were maintained under an argon atmosphere in a glove bag and the concentration in N2H4 in the feed solution was chosen as a function of the flow rate (1–5 mL·min−1) and the decomposition rate of N2H4 at 275 °C to compensate for the decomposed hydrazine amount.
2.8 Analytical
N2H4 and \( {\text{NH}}_{4}^{ + } \) (the by-product of thermal hydrazine decomposition) were analyzed by spectrocolorimetry with a Spectroquant® Nova60. The measurements were made after reaction of the sample with an aldehyde–dimethylamino-4-benzoïc reactant for hydrazine and phenol + hypochlorite salt for ammonium. The reactants were supplied as test kits by Merck©. The hydrazine concentration can be analyzed from 0.005 to 2.00 ± 0.002 mg·kg−1, and NH +4 from 0.010 to 3 ± 0.002 mg·kg−1.
Morpholine and ethanolamine were analyzed by cationic liquid chromatography (Dionex®, DX 120) in 0.01 mol·kg−1 H2SO4, or by electrophoresis (ACE-MDQ Beckman–Coulter) in a 75 µm thin capillary coupled with a 200 nm detector. The uncertainty of the method is typically 10 % but can be higher for ethanolamine when traces of alkali are present in the solution.
2.9 Thermodynamic Modelling
The prediction of in situ pH values, when required, was calculated from the chemical speciation of the fluid using the geochemical simulator software package JChess [27] and its associated database (version 2.0, release 2). JChess solves the set of equations involved in aqueous chemistry. At each step, the program distributes each element of the solution in stable aqueous species, calculates the activity coefficients of water and the aqueous species, the fugacity of gas, and the saturation index of the solution with respect to all of the phases of the database. The database was complemented with the morpholine/morpholinium and ethanolamine/ethanolamonium reactions according Eqs. 5 and 6. We used interpolation from measured values for morpholine [10] and ethanolamine [11].
3 Results and Discussion
3.1 Decomposition Rate of Hydrazine
The decomposition rate of hydrazine has been measured at 275 °C by monitoring the N2H4 concentration with time (and its by-product \( {\text{NH}}_{4}^{ + } \)) in solutions containing between 2 and 10 mg·kg−1 initially. These experiments were performed in the presence of morpholine or ethanolamine, according Table 2, to buffer the pH value around 6 at 275 °C. Complementary experiments where conducted with 1 g·L−1 magnetite added in the reactor, or with solutions of same pH but without amine. The details of the runs are presented in Table 3.
The decrease of hydrazine concentration measured during these tests is shown in Fig. 3 by plotting the logarithm of the relative decay against time. The concomitant increase of ammonia (not shown) confirms that this decrease is a consequence of the thermal decomposition of hydrazine. The linearity of the relationships is consistent with an expected first-order kinetic law, as expected from a disproportionation reaction ([19] and references therein). The rate constants of the first-order reactions are indicated in the figure.

Decrease of the hydrazine concentration with time. The constants refer to a first-order kinetic formalism
These results show the catalytic effect of magnetite, already suggested in the literature [17]. The results with morpholine show the variability of tests conducted with the same material, at the same pH and the same temperature. However, this does not prevent the conclusion that the decomposition rate is higher in the presence of the solids. A previous study [19] has pointed out the importance of the material in contact with the solution (titanium, Teflon and Ni-base alloy), as well as the cleaning method of the vessel (chemical or mechanical) on the thermal decomposition rate. Our measured decomposition rates in contact with titanium (or TiO2 passive layer) are two orders of magnitude slower than rates published for a similar temperature, but carried out in carbon steel or stainless steel vessels [16, 19]. These features indicate that the decomposition is a catalyzed reaction and may explain the differences observed here, since the cleaning of the vessel before each run may differ from run to run.
In addition to catalysis by solid surfaces, the possible effect of other aqueous components, such as amines type, has been tested but the run conducted without an amine is not significantly different from the others (Fig. 3). pH is another major factor influencing the decomposition kinetics. In Fig. 4 we compare our data with measurements at lower pH previously published [19] at 270 °C in the same titanium reactor. Despite the variability mentioned above, a clear influence of pH is observed and can be attributed to evolution of hydrazine species. The hydrazine speciation calculated at 275 °C using the acidity constant published by Cohen [9] is shown in the same figure, and the agreement between the relative proportion of N2H4 and \( {\text{N}}_{2} {\text{H}}_{5}^{ + } \) and the measured rates suggests that \( {\text{N}}_{2} {\text{H}}_{5}^{ + } \) has a slower decomposition rate than N2H4. Keeping in mind that the decomposition reaction is likely to be a catalyzed reaction, an alternative explanation is that the positive charge of the hydrazinium cation prevents its sorption onto the vessel, which is also likely to be positively charged at low pH (the pH corresponding to the point of zero charge, or pHZPC, from which the surface charge begins to be reversed is 6.3 at 25 °C for rutile [32], and is expected to be lower at 275 °C [33]).

Variation of the hydrazine thermal decomposition rate at 275 °C with pH, compared to the hydrazine speciation. The relative proportion of hydrazine and hydrazinium were calculated using the acidity constant reported in Cohen [8]
3.2 Stability of the Amines
The stability of the amines, tested in dedicated runs, is shown in Fig. 5. Ethanolamine appears stable over a month. Morpholine is decomposed in a few days in contact with magnetite but decomposes at a much slower rate in clear water. In any case, both amines are stable during the short residence times of these experiments.

Stability of morphokine and ethanolamine on time at 275 °C in a closed system
3.3 In Situ pH and Eh of Hydrazine–Morpholine–Ethanolamine Bearing Solutions at 275 °C
The reproduction, in the laboratory, of the PWR secondary circuit condition with amines, ammonia and hydrazine, in particular with a steady state hydrazine concentration in the range of 20–100 μg·kg−1, is a difficult task. For hydrazine, the feed solution reasonably balanced the thermal decomposition inside the reactor, as well as the possible oxidation. Its steady-state concentration was measured in the out-flowing solution and may be affected by the presence of oxygen traces in the feed solution, small variations in the flow rate of the pump, etc., and was not strictly constant over a run. Sometimes, the under- or over-evaluation of the starting N2H4 concentration led to inadequate hydrazine concentrations but pH and Eh values were systematically recorded up to 12 times per run. The results are presented in Table 4. This variability does not affect, however, the conclusions of the study, which are based on the measured values of concentrations, pH and Eh, and not on the predictions used for the dimensioning of the runs.
3.3.1 In Situ pH Determined from Potentiometric Measurements
The in situ pH measurements at 275 °C of both “low pH” and “high pH” conditions ranged respectively between 6.0 and 7.0 in morpholine bearing solution and between 5.5 and 7.3 in ethanolamine bearing solution. The variability of the pH data reflects variations in the concentration in KOH during the runs; some runs were conducted with KOH in both starting and feed solutions, others with KOH only in the starting solution, still others without KOH.
We compared our measured pH values with thermodynamic predictions. In pure water, the change of pH with temperature depends on the variation of the dissociation constant of water (pK w) only and can be easily calculated. This is illustrated in Fig. 6 where the change of pH with temperature of a pure hydrothermal solution is shown by grey lines. Briefly, the proton concentration remains constant in the acid range, where [H+] is higher than [OH−], leading to a pH which does not depend on temperature (vertical gray lines). In the alkaline region [OH−] is higher than [H+], the hydroxide ions concentration remains constant but pH decreases following the decrease of pK w (oblique grey lines). The near neutral region is more difficult to describe but this figure shows that the in situ pH in solutions at any temperature may basically be estimated from pH measurements at 25 °C. However, the accuracy of the prediction is lower in the presence of amines since the variation of their acidity constants with temperature should be taken into account. Moreover, thermodynamic calculations including the pKs of all of the aqueous species of the secondary circuit showed that a small error in the 8–9 pH25 °C range (degassing, carbonation) leads significant differences at high temperature. To give a detailed account, at 25 °C the pH of the amine-bearing solutions is around 9–9.5 and both the neutral and protonated species of morpholine or ethanolamine are present. At 275 °C the acidity constants of the amines are close to the neutral pH leading to their deprotonation during the temperature increase. Therefore, considering a small difference of the pH at 25 °C, the amount of released protons during the temperature change is important because of the sharp pH-speciation relationship in this 25 °C pH range. This is illustrated by three arrows in Fig. 6. An alternative method is to estimate the in situ pH by computing the speciation of the fluid at 275 °C, taking into accounts all of the aqueous species. For these calculations the measured concentrations of ammonia (initially present + produced by the hydrazine decomposition) was used, with the assumption that the amine concentration was constant during the runs and we calculated the concentration of KOH in the reactor (when added) at each time as a function of the starting and feed solution compositions.

Schematic variation of pH with temperature change in the presence of an amine
The thermodynamic predictions and the measurements are compared in Fig. 7 for two runs which yielded the most reliable data (stable electrodes, mixed-flow system). The observed differences do not exceed 0.2 pH units and therefore are not considered as being significant. Therefore, our results are in agreement with the previous measurements of the acidity constant as a function of temperature of morpholine [10] and ethanolamine [11].

Comparison between predicted and measured in situ pH in two representative runs
3.3.2 Redox Potential Imposed by Hydrazine
Recording the redox potential over time, in the mixed flow experiments, produced steady-state values at ±0 mV, provided that the concentration of hydrazine was higher than a few tenths of ppb. This condition was obtained with 0.6 mg·kg−1 N2H4 in the feed solution with 1.1–5 mL·min−1 flow rate. Below this threshold concentration in the vessel, the Eh values were erratic. The batch experiments devoted to the thermal decomposition of hydrazine are another source of Eh data. Although the hydrazine concentrations in these runs were much higher than in the mixed flow runs, the measured Eh values were of the same order of magnitude. The presence of magnetite did not seem to have an influence. An example is given in Fig. 8 where all of the available data for the morpholine conditioning are reported together. The dispersion of the data in this figure is higher than in a single experiment. The figure also suggests that the potential may be lower for higher N2H4 concentrations. The low measured potential is consistent with the one previous corrosion experimental study [14], conducted at 270 °C but with a different fluid chemistry based on Na2SO4 and NH4OH.

Record at 275 °C of the redox potential in the simulated conditioning fluids with morpholine. Mixed flow experiments are complemented with batch data from the decomposition experiments
Within the 0.4–1 mg·kg−1 N2H4 range, the pH appears to be a first-order parameter. This is illustrated in Fig. 8 where all of the data corresponding to this N2H4 range, obtained in batch experiments or in mixed flow runs with morpholine or ethanolamine, are plotted in a single Eh–pH diagram. The observed relation suggests that H+ plays a role in the redox potential when controlled by N2H4. Assuming that N2H4 is the sole electron donor in the system, the redox potential control likely results from the oxidation of hydrazine into N2 by the transfer of two electrons per nitrogen atom. This reaction consumes OH− according to Eq. 1, or equivalently generates protons though the water dissociation reaction.
The redox potential can also be predicted by thermodynamic considerations. The Nernst formalism applied to reaction 1 gives Eq. 7:
where T is the temperature in K, n the number of transferred electrons and F the Faraday constant. The dissociation constant of water, pK w, is 11.2 at 275 °C [20]. E stands for the in situ potential, E 0 for the standard potential at 275 °C. [H2O], [N2] and [N2H4] are the activities of water and the corresponding aqueous species.
Several difficulties appear in the calculation of E. First, the activity quotient is calculated from the concentration quotient, the activity coefficients of the neutral species being close to 1 in such dilute solution. By contrast with N2H4, the concentration of aqueous N2 is not measured and should be calculated. Actually, there are two possible sources of N2 with different stoichiometry: (1) the oxidation of N2H4 by traces of oxygen present in the feed solution and (2) the thermal decomposition. The relative contribution of these two reactions to the production of N2 was evaluated from the increase of aqueous NH3, produced only by the thermal decomposition, in order to obtain the stoichiometry of the budget reaction. The oxidation of N2H4 accounts for 42–93 % of the produced N2. However, the logarithm of the activity quotient is small and has a limited influence of the final potential. By contrast, the standard potential is a first order factor influencing the calculation of E. Values for E 0 at high temperature were previously reported by Lee [12] by considering the fugacity of gaseous N2 in the budget reaction. But the origin of the values, −1.21 V at 100 °C and −1.68 V at 288 °C, is not clearly explained (it is unclear whether these are measured or come from another source) and difficult to compare with our measurement using aqueous N2. We propose here, based on our own in situ potentiometric measurements, the value of −1.1 V for the standard potential of the reaction considering aqueous N2. The corresponding Eh–pH relationship is indicated on Fig. 9. Despite the complexity of the measurements and the calculations, this estimation is in agreement with the unique measurement in PWR where a potential around −0.5 V was given [14].

Eh–pH relationships in the experiments (batch or mixed flow) containing 0.04 mg·kg−1 < N2H4 < 1 mg·kg−1
4 Conclusions
Using a specific experimental mixed flow system to perform measurements in temperature and pressure conditions close to secondary circuits of PWR, new data on hydrazine chemistry have been obtained. First, no effect of amines used to control the pH (morpholine or ethanolamine) has been found on the rate of thermal decomposition. However, the presence of magnetite leads to a higher rate, consistent with a reaction catalyzed by metal oxides surfaces. The effect of pH has also been investigated, showing a higher stability for hydrazinium in comparison with neutral hydrazine species. The use of high temperature electrodes has allowed the measurement of pH and Eh of simulated secondary circuit solutions. A Nernstian behavior of measured potential versus pH has been found. Although the redox couple at the origin of the measured Eh remains difficult to identify, we suggest the oxidation of hydrazine with production of aqueous N2, has a standard potential of −1.1 V at 275 °C.
The redox and acid–base properties of a simulated secondary circuit conditioning determined in this experimental study may help to better predict the fluid chemistry under operating conditions. In particular, the experimental device developed here may be used to test other possible conditions. Another application of this instrumentation is its use to follow on-line the evolution of pH and Eh in cooling circuits. Several works in this field were performed previously (Niedrach [23], Lvov et al. [25]), leading to the development of several innovative systems. However, this instrumentation is not yet used in nuclear power plants, where an analysis of cooled aliquots is the usual protocol. However, this approach is based on the knowledge of the relation between high temperature–high pressure characteristics of the solution and the data measured at room temperature (mainly concentration of amines and pH). In-situ and real-time data on pH and Eh could help one to react more rapidly to the evolution of these key parameters in comparison to analysis of aliquots, and to ensure the validity of the relation between room temperature analysis and characteristics of cooling solutions from high temperature.
References
Brandes, J., Boctor, N., Cody, G., Cooper, B., Hazen, R., Yoder, H.: Abiotic nitrogen reduction on the early Earth. Nature 395, 365–367 (1998)
Edmond, J., von Damm, K., McDuff, R., Measures, C.: Chemistry of hot springs on the east Pacific rise and their effluent dispersal. Nature 297, 187–191 (1982)
Ferris, J.P.: Chemical markers of prebiotic chemistry in hydrothermal systems. Orig. Life Evol. Biosph. 22, 109–134 (1992)
Ferris, J.P.: The potential for prebiotic synthesis in hydrothermal systems. Orig. Life Evol. Biosph. 24, 363–381 (1994)
Takano, Y., Sato, R., Kaneko, T., Kobayashi, K., Marumo, K.: Biological origin for amino acids in a deep subterranean hydrothermal vent, Toyoha mine, Hokkaido, Japan. Org. Geochem. 34, 1491–1496 (2003)
Stephen-Sherwood, E., Oro, J., Kimball, A.P.: Thymine: a possible pre-biotic synthesis. Science 173, 446–447 (1971)
Audrieth, L.F., Ogg, B.A.: The Chemistry of Hydrazine. Wiley, New York (1951)
Cohen, P.: The ASME Handbook on Water Technology for Thermal Power Systems. American Society of Mechanical Engineers, New York (1989)
Hitch, B.F., Mesmer, R.E.: The ionization of aqueous ammonia to 300 °C in KCl media. J. Solution Chem. 5, 667–680 (1976)
Ridley, M.K., Xiao, C., Palmer, D.A., Wesolowski, D.: Thermodynamic properties of the ionization of morpholine as a function of temperature and ionic strength. J. Chem. Eng. Data 45, 502–507 (2000)
Bénézeth, P., Wesolowski, D.J., Palmer, D.A.: Potentiometric study of the dissociation quotients of aqueous ethanolammonium ion as a function of temperature and ionic strength. J. Chem. Eng. Data 48, 171–175 (2003)
Lee, J.B.: Electrochemical approach to the corrosion problems of several iron–nickel–chromium alloys in high temperature high pressure water. PhD thesis, Ohio State University, USA (1978)
Charlot, G.: L’analyse qualitative et les réactions en solution. Masson et Cie, librairies de l’académie de médecine (1957)
Stellwag, B., Killian, R.: Influence of O2 and N2H4 on the ECP in high temperature water. In: Proceedings of the 5th International Symposium on Environmental Degradation of Materials in Nuclear Power Systems–Water Reactors, pp. 96–105 (1991)
Baker, M.D., Marcy, V.M.: Hydrazine as an oxygen scavenger—A progress report on tests at Springdale Station. Trans. ASME 78, 299–304 (1956)
Dickinson, N. L., Felgar, D.N., Pirsh, E.A.: An experimental investigation of hydrazine–oxygen reaction rates in boiler feedwater. In: Proceedings of the American Power Conference vol. 19, p. 692 (1957)
Resch, G.: Über das Verhalten von Hydrazinhydrat unter den Bedingungen des Hochdruckkessel betriebes, Hochschule Braunschweig, Thesis, (1957)
Hader, K.: Thermische Zersetzung von Hydrazin inwässriger Lösung unter Betriebsbedingungen eines Kraftwerkes. Goerg Simon Ohm-Fachhochschule Nürnberg (thesis) (1987)
Delaunay, S., Mansour, C., Pavageau, E. M., Berger, G., Cote, G., Lefevre, G., Federoff, M.: Redox properties and stability of hydrazine in the context of the secondary circuits of PWR plants. Nuclear Plant Chemistry Conference (2010)
Mesmer, R.E., Baes Jr, C.F., Sweeton, F.H.: Acidity measurements at elevated temperatures: IV. Apparent dissociation of water in 1 m potassium chloride up to 292 °C. J. Phys. Chem. 74, 1937–1954 (1970)
Mesmer, R.E., Palmer, D.A., Wesolowski, D.J.: Potentiometric studies at ORNL with hydrogen electrode concentration cells. Physical Chemistry of Aqueous Systems: Meeting the Needs of Industry. Begell House, New York (1995)
Wesolowski D.J., Palmer, D.A., Mesmer, R.E.: Measurements and control of pH in hydrothermal solutions. Water–Rock Interaction, Balkema, pp. 51–55 (1995)
Niedrach, L.W.: A new membrane-type pH sensor for use in high temperature—high pressure water. J. Electrochem. Soc. 127, 2122–2130 (1980)
Macdonald, D.D., Hettiarachchi, S., Lenhart, S.J.: The thermodynamic viability of yttria-stabilized zirconia pH sensors for high temperature aqueous solutions. J. Solution Chem. 17, 719–732 (1988)
Lvov, S.N., Zhou, X.Y., Ulmer, G.C., Barnes, H.L., Macdonald, D.D., Ulyanov, S.M., Benning, L.G., Grandstaff, D.E., Manna, M., Vicenz, E.: Progress on yttria-stabilized zirconia sensors for hydrothermal pH measurements. Chem. Geol. 198, 141–162 (2003)
Lefevre, G., Berger, G.: Potentiometry up to 275 °C: example of pH titrations of cobalt ferrite particles. J. Colliods Interface Sci. 430, 12–17 (2014)
Van der Lee, J., De Windt, L.: CHESS Tutorial and Cookbook. Users Manuals Nr. LHM/RD/02/13. Ecole des Mines de Paris, Fontainebleau, France (2002)
Macdonald, D.D., Hettiarachchi, S., Song, H., Makela, K., Emerson, R., Ben-Haim, M.: Measurement of pH in subcritical and supercritical aqueous systems. J. Solution Chem. 21, 849–881 (1992)
Bosch, R.W., Bogaerts, W.F., Zheng, J.H.: Simple and robust external reference electrode for high temperature electrochemical measurements. Corrosion 59, 162–171 (2003)
Mansour, C., Lefèvre, G., Pavageau, E.-M., Catalette, H., Fédoroff, M., Zanna, S.: Sorption of sulfate ions onto magnetite. J. Colloids Interface Sci. 331, 77–82 (2009)
Barale, M., Mansour, C., Carrette, F., Pavageau, E.-M., Catalette, H., Lefèvre, G., Fedoroff, M., Cote, G.: Characterization of the surface charge of oxide particles of PWR primary water circuits from 5 to 320 °C. J. Nucl. Mater. 381, 302–308 (2008)
Villasenor Nano, G., Strathmann, T.J.: Ferrous iron sorption by hydrous metal oxides. J. Colloids Interface Sci. 297, 443–554 (2006)
Ridley, M.K., Machesky, M.L., Welelowski, D.J., Palmer, D.A.: Calcium adsorption at the rutile–water interface: a potentiometric study in NaCl media to 250 °C. Geochim. Cosmochim. Acta 63, 3087–3096 (1999)
Acknowledgments
The authors thank Didier Noël (EDF) and Laurent Truche (Université Nancy) for fruitful discussions on Soret effect and on the redox reactions, and the helpful comments of two anonymous reviewers. We thank EDF for its financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Million-Picallion, L., Berger, G., Lefèvre, G. et al. Stability of Hydrazine, Morpholine and Ethanolamine at 275 °C and In Situ Measurement of Redox and Acid–Base Properties. J Solution Chem 44, 1900–1919 (2015). https://doi.org/10.1007/s10953-015-0375-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10953-015-0375-4