
Abstract
(La0.8Ca0.2)1−xBixFeO3 (x = 0.0, 0.1, and 0.2) compounds were prepared by the sol-gel method, using the citric acid route. Structural and microstructural characteristics of the samples were studied by X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), and Raman spectroscopy. Mössbauer spectroscopy was also used for magnetic characterization and structural confirmation of X-ray data. Refinement of XRD patterns indicated that the compounds crystallize in an orthorhombic structure, Pnma space group. The SEM measurements showed that the morphology of the samples is characterized by the existence of large agglomerates, while TEM confirmed the fine particle size. Raman spectra revealed that, when increasing temperature, the level of oxygen species adsorbed at the surface of the particles increases progressively. Room temperature Mössbauer spectroscopy data revealed the existence of two octahedral magnetic sites in the structure of the compounds, which is compatible with XRD data, and a small percentage of a paramagnetic phase in the samples, which increases with Bi-content.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Recently, there has been a growing interest in the study of layered perovskites of general formula ABO3 (A, B = metals), which show a broad range of attractive physical properties, leading to a wide variety of applications [1, 2]. In this context, researchers have been particularly interested to the study of bismuth ferrite, BiFeO3, which is a ferroelectric TC = 1100 K (Curie temperature) multifunctional material that shows a magnetic ordering at TN = 640 K (Néel temperature) [3,4,5,6,7]. Bismuth ferrite is indeed one of the most interesting multiferroic materials, thanks to its excellent properties for technological applications (sensors, ultrafast optoelectronic devices, multistate memory devices, etc. [8,9,10]).
Mössbauer spectroscopy has been used very successfully to investigate this type of materials. For example, investigation of doped LaFeO3 by this technique has yielded particularly interesting results, one of which is the finding that La1−xSrxFeO3 shows an isomeric shift between the Fe3+ and Fe4+ characteristic regions, which has been taken as an evidence of an intermediate state valence between Fe3+ and Fe4+ in this material [11]. On the other hand, the observed intermediate valence iron state is the result of the electron jump between the paired Fe3+ and Fe4+ ions, as previously observed for Fe2+ions and Fe3+ in the octahedral sites of magnetite [12, 13]. Also, Benali et al. investigated a series of compounds with general formula La0.8Ca0.2−xPbxFeO3 (0.0 < x < 0.2) using Mössbauer spectroscopy [14], concluding on the presence of Fe3+ tetrahedral sites, which increase in number with the lead content.
In order to contribute to the exploration of the structural and physical properties of those mixed perovskite-like materials, in the present work, we investigated the effect degree of substitution of lanthanum and calcium by bismuth on the structural, morphological, and optical properties of compounds belonging to the (La0.8Ca0.2)1−xBixFeO3 (x = 0.0, 0.1, and 0.2) series. These materials were prepared by the sol-gel method, and Mössbauer spectroscopy was chosen in the present work as one of the main techniques to use in the investigation of the studied materials, together with Raman spectroscopy, X-ray diffraction, and microscopy techniques.
2 Experimental
2.1 Synthesis
The (La0.8Ca\(_{\mathrm {0.2}})_{\mathrm {1-}_{x}}\)BixFeO3 (x = 0.0, 0.1, and 0.2) compounds were synthesized by the sol-gel method [15], using lanthanum nitrate, bismuth nitrate, calcium nitrate, ferric nitrate, and citric acid, according to the following reaction equation:
In a first step, the precursors La(NO3)3,Ca(NO3)2, Bi(NO3)2, and Fe(NO3)3 were mixed in ion-free water. All precursors used were nitrates with 99.9% purity. After that, citric acid and ethylene glycol were added under constant stirring at room temperature for 5 h, to improve the consistency and consequent formation of the gel. The obtained mixture was then heated at 170 °C until a black powder was formed. This powder was pressed into thin pellets of ca. 12 mm in diameter and ca. 1.5 mm in thickness and submitted to heat treatments at different temperatures (600 and 700 °C for 12 h; 800 °C for 72 h), interrupted by grinding cycles. Finally, the powder was sintered at 900 °C for 72 h and slowly cooled to room temperature. This method was chosen because it is known to yield samples with a high degree of purity [10].
2.2 Apparatus
The phase purity, homogeneity, lattice structure, and cell parameters of the synthesized compounds were checked by X-ray diffraction (XRD) analysis, using a Burker 8D Advance X-ray powder diffractometer, with CuKα1 radiation (λ = 1.5406 Å), in θ–2θ Bragg-Brentano geometry. The acquisition was in the 2θ range of 10–100°, with a step of 0.02° and an acquisition time for each step of 1 s. The XRD data were also used for obtaining the lattice parameters by means of Rietveld analysis, using the FULLPROF program.
Magnetic measurements were done in a vibrating sample magnetometer (VSM) with a cryogen-free Dynacool PPMS, operating at a vibration frequency of 40 Hz and amplitude of 2 mm in the central area of the coils. The samples were in a rod-shaped sample holder made of Perspex.
Differential scanning calorimetry (DSC) was performed using a PerkinElmer STA6000 system, with a step of 20 °C min− 1.
Raman spectra were obtained for the different samples, in the wavenumber range 50–1500 cm− 1, using a Raman micro-system (Horiba LabRam HR Evolution), equipped with a Synapse CCD detector, a high-stability BXFM open-space confocal microscope, and a 600 g mm− 1 grating, with 532 nm excitation. The laser power at the sample was approximately 17 mW, and the exposure time 30 s. A 50× objective lens was used. The final spectra are the mean of 60 scans for every sample.
Mössbauer spectra were recorded at room temperature by a constant acceleration-type spectrometer (WissEL) in transmission geometry, using the 57Fe as isotope and a source of 57Co (energy of 14.4 keV) in a matrix of Rh with an activity of about 20 mCi. The spectra were fitted by a set of Lorentzian lines determined by the least squares method, applying the NORMOS program distributed by WissEL GmbH. Isomer shifts are given, as usually, relatively to α-Fe measured at room temperature.
Microstructures were observed by a TESCAN VEGA3 SBH scanning electron microscope, operating at 20 kV and equipped with an EDS detector BurkerXFlagh 410M, which allowed the detection of the characteristic X-rays emitted by the sample and, consequently, the identification of its chemical elements. For the analysis, a small amount of the powder of the compounds was deposited on a carbon tape.
The transmission electron microscopy (TEM) allowed the observation of the particles morphology and determination of their average size. Moreover, this technique provided information on the crystalline phases of the materials by obtaining electron diffraction patterns. TEM images were taken with a FEI Tecnai G2 with an acceleration voltage of 200 kV, in bright field and electronic diffraction modes. For the analysis, the carbon grids were immersed in dilute suspensions of the powders and, after, the solvent evaporation the grids were placed in the microscope. For the treatment of the obtained images and to obtain the size of the particles, we used the ImageJ software.
X-ray fluorescence (XRF) analysis was done to confirm the chemical elements present in the samples. A Hitachi SEA6000VX benchtop high-sensitivity analyzer was used equipped with an X-ray tube with a tungsten target, operating at potentials of 15 and 50 kV and a current of 1000 mA, and a 3 mm-wide primary-beam collimator. The SEA6000VX measurement geometry is fixed, with its energy-dispersive Vortex Si semiconductor detector positioned at a scattering angle of 135° and a distance of 19 mm from the specimen, with no additional slits to restrict its acceptance angle.
3 Results and Discussion
3.1 X-Ray Diffraction, X-Ray Fluorescence, Scanning Electron Microscopy, and Transmission Electron Microscopy
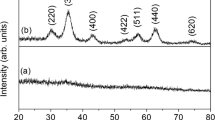
The room temperature X-ray diffraction patterns for the studied (La0.8Ca0.2)1−xBixFeO3 (x = 0.0, 0.1, and 0.2) compounds are shown in Fig. 1. These data show that these compounds exhibit a single perovskite phase, at least within the detection limits of the instrument. Good crystallinity is observed since the peaks of the diffractograms are narrow and intense.

Room temperature XRD patterns for (La\(_{\mathrm {0.8}}\)Ca\(_{\mathrm {0.2}})_{\mathrm {1-}_{x}}\)BixFeO3 (x = 0.0, 0.1, and 0.2) compounds
The performed Rietveld refinements of the XRD patterns revealed that the studied compounds crystallize in an orthorhombic structure, belonging to the Pnma space group. Table 1 shows the obtained structural parameters.
The average crystallite sizes (Dsc) (see Table 1) were also estimated from the XRD patterns, using the Scherer formula [16]:
where λ is the used wavelength, θ is the Bragg angle for the most intense peak, and β is the half height width of this peak.
Figure 2a shows the variation of the lattice parameters as a function of ion radii. The mean ion radii of the cations occupying the A-site, <rA>, were determined from the following relationship:
The substitution of the larger calcium ion (\(r_{\text {Ca}}^{\mathrm {2+}}\) = 1.18 Å) and lanthanum ion (\(r_{\text {La}}^{\mathrm {3+}}\) = 1.216 Å) by the smaller bismuth ion (\(r_{\text {Bi}}^{\mathrm {3+}}\) = 1.03 Å) induced a decrease of the average ion radii <rA>, but unit cell parameters and unit cell volume increase (Fig. 2b). There are several factors responsible for the differences in the values of cell parameters and cell volume that are difficult to establish. In this case, <rA>is not the only parameter with influence in the structure. This was observed in other perovskites and attributed to an increase of the angles between the sites [17].

Variation of the unit cell parameters (a–c) as a function of the ion radii of the cations occupying the A-site (a), the unit cell volume (V ) (b), and the mean crystallite size (Dsc) as a function of bismuth content in the samples (c). The line is a guide to the eyes
The surface morphology of the studied materials was estimated by scanning electron microscopy (SEM). Figure 3 shows the obtained room temperature SEM micrographs of the (La0.8Ca0.2)1−xBixFeO3 (x = 0.0, 0.1, and 0.2) compounds. These figures show the presence of a large distribution of grain and illustrate a sample with less voids and densified; also, it can be seen that all particles are in spherical form. The TEM images have been used to investigate the distribution of the particle size and evaluate the degree of agglomeration of crystallites. Figure 4a–c proves examples of the images and selected area electron diffraction (SAED) patterns/particle size distribution of the (La0.8Ca0.2)1−xBixFeO3 (x = 0.0, 0.1, and 0.2) compounds, respectively. Manual statistical particle size counting was executed on TEM images using Image-J software.

SEM micrographs of (La\(_{\mathrm {0.8}}\)Ca\(_{\mathrm {0.2}})_{\mathrm {1-}_{x}}\)BixFeO3 (x = 0.0, 0.1, and 0.2) compounds

a–c TEM images and SAED/particle size distributions (insets) of (La0.8Ca0.2)1−xBixFeO3 (x = 0.0, 0.1, and 0.2) compounds
The size of particles is distributed accordingly to a Lorentzian law, for samples x = 0.0 and 0.1. The particle size distribution is random for the x = 0.2 sample. If we compare these values by those of the sizes of crystallites determined by the Scherer formula relating to X-ray diffraction diagrams, we find that these are the largest, being the particle size of about four times the crystallite size, suggesting that each grain observed is formed by several crystallites. This has also been observed in the case of many other materials [18]. The estimated average particle sizes are of the order of 284.1 ± 56.8, 291.8 ± 55.6, and 272.8 ± 89.4 nm, respectively (see Table 1).
The SAED patterns of the compounds, composed by circles and/or small dots, confirm their crystallinity, in agreement with the diffractograms obtained by XRD.
Both XRF and EDS analyses confirmed the presence of all expected elements, lanthanum (La) bismuth (Bi), calcium (Ca), iron (Fe), which confirms that they are all non-volatile and that during synthesis, there was no loss of any integrated element (see Figs. 5 and 6).

EDS spectra of (La0.8Ca0.2)1−xBixFeO3 (x = 0.0, 0.1, and 0.2) compounds

XRF spectra of (La0.8Ca0.2)1−xBixFeO3 (x = 0.0, 0.1, and 0.2) compounds
3.2 Magnetic (VSM) and Calorimetry (DSC) Results
Figure 7 shows the temperature dependence of magnetization (M–T curve), measured under a field of 500 Oe and in the temperature range of 1.8–300 K in the ZFC and FC modes.

Field cooled (FC) and zero field cooled (ZFC) curves measured at 500 Oe for (La0.8Ca0.2)1−xBixFeO3 (x = 0.0, 0.1, and 0.2) compounds
As one can see, the insertion of bismuth ions widely affect the magnetization. Without bismuth, the ZFC magnetization curve shows two clear maxima at Tmax1 = 13 K and Tmax2 = 106 K, and FC curves deviate significantly from the ZFC ones below Tmax2.
This transition temperature is in the range of TN = 100–110 K, reported elsewhere [19,20,21,22]. The second maximum temperature can be attributed to ferromagnetic phase that coexists with the main antiferromagnetic one, or the antiferromagnetic state is frustrated.
One can see that the insertion of 10% of bismuth ions increases the temperature of the first maximum in the ZFC curve (Tmax1 = 60 K) with the appearance of a maximum in the FC curve at the same temperature. This first transition temperature disappeared totally when introducing 20% of bismuth. As it is known, when substituted 20% of Lanthanum with calcium ions there is a competition between ferromagnetism and antiferromagnetism between Fe3+ −Fe3+ (FM) and Fe3+ −Fe2+ (AFM). When introducing Bi3+ ions, the percentage of Fe3+ ions increases which increases the Fe3+ −Fe3+ interaction and automatically the enhancement of the magnetization, as one can see in Fig. 7.
The magnetic field dependence of the specific magnetization (M–H curve) obtained from VSM measurements for (La0.8Ca\(_{\mathrm {0.2}})_{\mathrm {1-}_{x}}\)BixFeO3 nanoparticles measured at 2 K and room temperature (RT) with magnetic field (H) in the range of 9 Tesla are shown in Fig. 8.

Magnetic field dependence of the specific magnetization (M–H curves) obtained for (La0.8Ca0.2)1−xBixFeO3 (x = 0.0, 0.1, and 0.2) compounds measured at 2 K and room temperature (RT)
All the samples exhibit weak ferromagnetic behavior (unsaturated curves) and increase of magnetization (M) with increased content of bismuth concentration. The sample with x = 0.2 shows the highest M value of 0.166 emu g− 1.
One can see in the hysteresis loops recorded at 2 K the existence of weak ferromagnetism contribution, but the antiferromagnetic one dominates the magnetization in all compounds. The insertion of bismuth ions act to decrease the coercivity (Hc) and the remanent magnetization (Mr).
In these types of perovskites, a ferroparamagnetic transition is expected between 600 and 700 K [14]. This transition is shown for our samples by DSC measurements in Fig. 9. The transitions are observed by the derivative of heat flow and are about 392–393 °C.

DSC curves and their derivatives for (La0.8Ca0.2)1−xBixFeO3 (x = 0.0, 0.1, and 0.2) compounds
3.3 Raman Spectroscopy Results
The Raman spectra of the samples shown in Fig. 10 were found to be sensitive to the sample history, in particular to the level of adsorbed oxygen species, as noticed by the observation of the characteristic band at ca. 775 cm− 1 in some of the samples. For the La0.8Ca0.2FeO3 parent sample (x = 0.00), the peak characteristics of the LaFeO3 reported in the literature dominate the spectrum: observed at 150, 174, 255, 288, 410, 430, and 652 cm− 1 in the studied sample, matching well the previously reported values (145, 170, 255, 285, 400, 430, and 640 cm− 1 [18, 23, 24]). Other bands were observed at 98, 131, 1121, and 1315 cm− 1, which may also be due to LaFeO3 or result from the presence of calcium in the sample. Besides, this sample exhibits the above-mentioned intense band at 775 cm− 1 due to adsorbed oxygen species at the surface of the nanoparticles. The sample with the smallest amount of Bi-dopant (x = 0.1) has a room temperature Raman spectrum similar to that of the La0.8Ca0.2FeO3 sample (x = 0.0) with the following major differences:
-
i.
The sample is practically free of adsorbed oxygen species, as seen by the small intensity of the band at 775 cm− 1 in its Raman spectrum;
-
ii.
In the Raman spectrum of the x = 0.1 sample, the bands that act as indicators of the presence of the Bi-dopant, at ca. 554 and 1096 cm− 1 are already clearly observed.

Room temperature Raman spectra of (La0.8Ca0.2)1−xBixFeO3 (x = 0.0, 0.1, and 0.2) compounds
The sample with the highest amount of Bi-dopant (x = 0.2) shows a different Raman spectrum profile. The bands due to LaFeO3 appear broadened (this is clearly seen in the whole spectral range investigated, but particularly noticeable in the low-frequency region, between 50 and 200 cm− 1, which appear much less well resolved in the spectrum of this sample), while the bands indicating the presence of the Bi-dopant (at ca. 554 and 1096 cm− 1) dominate the spectrum.
The temperature dependence of the Raman spectrum for x = 0.1 found that, with the temperature enhancement, the level of oxygen species adsorbed at the surface of the nanoparticles increases progressively (Fig. 11). Preliminary temperature variation studies on the sample with x = 0.15 point to the same tendency also for this sample, thus suggesting this behavior is a general trend for all samples. Finally, the spectra get below and above the ferromagnetic to paramagnetic phase transition temperatures (ca. 387 °C) for x = 0.1 appear to be distinctive, in particular in what concerns the strong intensity reduction of the bands at ca. 410/430 and 1315 cm− 1, which appear to be good indicators of the phase transition in the studied materials.

Temperature variation Raman spectra for x = 0.1 sample
3.4 Mössbauer Spectroscopy Results
Mössbauer spectra of the samples were registered, in order to confirm the valence state of the iron ions and to study their environment.
The Mössbauer spectra of (La0.8Ca\(_{\mathrm {0.2}})_{\mathrm {1-}_{x}}\)BixFeO3 (x = 0.0, 0.1 and 0.2) samples, obtained at room temperature are shown in Fig. 12. The solid lines represent the fit of a set of Lorentzians to the experimental data. The hyperfine parameters obtained from the fit to spectra are illustrated in Table 2.

Room temperature Mössbauer spectra of (La0.8Ca0.2)1−xBixFeO3 (x = 0.0, 0.1, and 0.2) compounds and fitted curves describing the experimental data (S1 and S2, magnetic sub-spectra; D, doublet)
The Mössbauer spectra prove well-resolved peaks, being clear that the insertion of Bi3+ ions does not significantly change the shape of the Mössbauer spectra. All spectra were fitted with two octahedral Fe3+ sextets (QS < 0) and a small percentage of a doublet that can be due to the presence of some iron in a paramagnetic phase or to an iron-containing impurity phase, although impurities were not found by X-ray diffraction. The percentage of the doublet increases with increasing of Bi-content, being 1.6% for x = 0.0 and 8.3% for x = 0.20. This latter sample has also a random distribution of particle size, as seen by TEM. A great part of the particles of this sample has a size inferior to the average size (272.8 ± 89.4 nm).
The observed octahedral Fe3+ sites are in conformity with XRD results, and the existence of magnetic sextuplets in all spectra confirms the magnetic behavior of the samples at room temperature. The obtained hyperfine parameters (Table 2) are in agreement with the parameters obtained for the family of perovskites of the system Ba–Bi–Fe–O [25] and for BiSrFeO and BiCaFeO perovskites [26].
4 Conclusion
In the present research work, we have successfully synthesized, using the sol-gel method, a series of (La0.8Ca0.2)1−xBixFeO3 (x = 0.0, 0.1, and 0.2). The nature of the phases of these samples was investigated by XRD, EDX, XRF, SEM, TEM, Raman, and Mössbauer spectroscopies. Rietveld refinement of XRD data showed that all compounds crystallized in an orthorhombic structure within the Pnma space group. Mössbauer measurements indicated the existence of two octahedral Fe3+ magnetic sites in the structure, in accordance with XRD results. TEM images confirmed the crystallinity of compounds and showed particle sizes about 200 nm. The Raman spectra, of the different materials, were recorded and assigned, and some structure-sensitive band markers were identified. In addition, the temperature dependence of the Raman spectra revealed that the level of oxygen species adsorbed at the surface of the particles increases with temperature and showed also that the spectra obtained below and above the ferromagnetic to paramagnetic phase transition temperature (ca. 387 °C, for the sample with x = 0.1) appear to be distinctive, in particular in what concerns the strong intensity reduction of the bands at ca. 410/430 and 1315 cm− 1, which then appear to be good indicators of the phase transition in the studied materials.
References
McCarty, J.G., Wise, H.: Catal. Today 8, 231 (1990)
Tofan, C., Klvana, D., Kirchnerova, J.: Appl. Catal. B 36, 311 (2002)
Suresh, P., Srinath, S.: J. Alloys Compd. 554, 271 (2013)
Hou, Z.L., Zhou, H.F., Kong, L.B., Jin, H.B., Qi, X., Cao, M.S.: Mater. Lett. 84, 110 (2012)
Godara, S., Sinha, N., Kumar, B.: Ceram. Int. 42(1), 1782 (2016)
Pandit, P., Satapathy, S., Gupta, P.K.: Physica B 406(13), 2669 (2011)
Ramesh, R., Spaldin, N.A.: Nat. Mater. 6, 21 (2007)
Sosnowska, I., Neumaier, T.P., Steichele, E.: J. Phys. C: Solid State Phys. 15, 4835 (1982)
Cheong, S.W., Mostovoy, M.: Nat. Mater. 6, 13 (2007)
Guanghua, Y., Michel, S.: Ceram. Bull. 70, 1173 (1991)
Shimony, U., Knudsen, J.M.: Phys. Rev. 144, 361 (1966)
Gallagher, P.K., MacChesney, J.B.: Symp. Faraday Soc. 1, 40 (1968)
Bauminger, R., Cohen, S.G., Marinov, A., Ofer, S., Segal, E.: Phys. Rev. 122, 1447 (1961)
Benali, A., Bejar, M., Dhahri, E., Sajieddine, M., Graça, M.P.F., Valente, M.A.: Mater. Chem. Phys. 149, 467 (2015)
Graça, M.P.F., Nico, C., Peres, M., Valente, M.A., Monteiro, T.: J. Nanosci. Nanotechnol. 12, 1 (2012)
Guinier, A.: In: Dunod, X. (ed.) Théorie et Technique de la Radiocristallographie. 3rd, p 462 (1964)
T.A. Ho, N.T. Dang, T.L. Phan, D.S. Yang, B.W. Lee, S.C. Yu: Journal of Alloys and Compounds (in press), https://doi.org/10.1016/jallcom.2016.03.156 (2016)
Ungár, T., Tichy, G., Gubicza, J., Hellmig, R.J.: Powder Diffr 20, 366 (2005)
Bokov, V.A., Grigoryan, N.A., Bryzhnina, M.F.: Phys. Status Solidi 20, 745 (1967)
Garcia-Munoz, J.L., Frontera, C., Aranda, M., Llobet, A., Richter, C.: Phys. Rev. B 63, 064415 (2001)
Frontera, C., Garca-Muno, J.L., Llobet, A., Aranda, M.A.G., Ritter, C., Respaud, M., Vanacken, J.: J. Phys.: Condens. Matter 13, 1071 (2001)
Troyanchuk, I.O., Mantytskaya, O.S., Chobot, A.N.: Phys. Status Solidi 44, 2266 (2002)
Popa, M., Franti, J., Kakihana, M.: Solid State Ion. 154–155, 135 (2002)
Wang, Y., Zhu, J., Zhang, L., Yang, X., Lude, L., Wang, X.: Mater. Lett. 60, 1767 (2006)
Boullay, Ph., Hervieu, M., Nguyen, N., Raveau, B.: J. Solid-State Chemistry. 147, 45 (1999)
Lepoittevin, C., Malo, S., Barrier, N., Nguyen, N., Van Tendeloo, G., Hervieu, M.: J. Solid-State Chemistry. 181(10), 2601 (2008)
Funding
The Coimbra Chemistry Centre (CQC) is supported by FCT, through the project UI0313/QUI/2013, also co-funded by FEDER/COMPETE 2020-EU. This work was also supported by funds from FEDER (Programa OperacionalFactores de Competitividade COMPETE) and from FCT-Fundação para a Ciência e a Tecnologia under the Project No. UID/FIS/04564/2016. Access to TAIL-UC facility funded under QREN-Mais Centro Project No. ICT_2009_02_012_1890 is gratefully knowledge. We thanks Dr. Goestar Klingelhoefer from Johannes Gutenberg University, Mains, for having given the Mössbauer source.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Issaoui, H., Benali, A., Bejar, M. et al. Structural, Morphological, Raman, and Mössbauer Studies on (La0.8Ca0.2)1−xBixFeO3 (x = 0.0, 0.1, and 0.2) Compounds. J Supercond Nov Magn 32, 1571–1582 (2019). https://doi.org/10.1007/s10948-018-4850-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10948-018-4850-1