
Abstract
The structural, electronic, magnetic, and mechanical properties of CoVTiAs and CoVTiSi quaternary Heusler alloys are studied using the full-potential linearized augmented plane-wave (FP-LAPW) method in the framework of the density functional theory (DFT). The generalized gradient approximation (GGA) is chosen for the exchange-correlation energy, whereas the modified Becke-Johnson (mBJ) formalism is applied for the electronic properties. Both CoVTiAs and CoVTiSi are stable in the type 2 (FM) structure. The mechanical properties of CoVTiX (X = As, Si) are predicted from the calculated elastic constants. Results identify that these compounds are mechanically stable. Our results with the mBJ approximation predict that CoVTiAs and CoVTiSi alloys are half-metallic ferromagnets with band gaps of 0.450 and 0.424 eV, respectively.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
In recent years, Heusler alloys have attracted the attention of many researchers due to their uses in spintronic applications such as tunneling magnetoresistance (TMR) and giant magnetoresistance (GMR) [1]. There are two classes of Heusler alloys that include full- (FHA) and half-Heusler alloys (HHA). The FHA have L21 structure with X2YZ chemical composition forming four interpenetrating face-centered cubic (fcc) lattices, where X and Y are transition metals and Z is a main group element. However, the HHA has the C1b structure with the XYZ chemical composition forming three interpenetrating fcc lattices and one vacant lattice. Most of these alloys exhibit half-metallic electronic property with a semiconducting structure in only one spin direction, whereas the other spin channel is strongly metallic. Another category of Heusler compounds known as quaternary Heusler alloys with the chemical formula XX′YZ with LiMgPdSb-type crystal structure and \(F\overline {4} 3m\) seul F-43m space group [2, 3] have attracted a great deal of interest. Galanakis [4] predicted that Co2[Cr1−xMnx]Al, Co2Mn[Al1−xSnx], and [Fe1−xCox]2MnAl are typically half-metallic ferromagnets with total spin magnetic moments that vary linearly between 2 and 4 BμB for Fe2MnAl and Co2MnAl compounds, respectively. The substitutional series of the quaternary Heusler compound Co2Mn1−xFexSi [5] was synthesized and investigated both experimentally and theoretically. For example, Alijani et al. have performed theoretical and experimental investigations of NiFeMnGa, NiCoMnGa, and CuCoMnGa [6]. They found that NiFeMnGa and NiCoMnGa exhibit excellent half-metallic ferromagnetism, while CuCoMnGa was found to be a metallic ferromagnet. In addition, Goa et al. studied the electronic, structural, and magnetic properties of CoFeCrZ (Z = Al, Si, Ga, and Ge) [7] using full-potential linearized augmented plane-wave (FP-LAPW) method. They found that both CoFeCrGa and CoFeCrGe quaternary Heusler alloys are nearly half metals, while CoFeCrAl and CoFeCrSi are excellent half-metallic (HM) ferromagnets with gaps of 0.16 and 0.28 eV, respectively. Nehra et al. [8] also showed that CoFeCrAl exhibits a HM ferromagnetic structure with a band gap of 0.41 eV around the Fermi level in the minority spin channel. Experimentally, the quaternary equiatomic Heusler alloy (CoFeMnGe) was found to have the cubic Heusler structure of LiMgPdSn type with a considerable amount of DO3 disorder at Curie temperature of 750 K [9]. Using FP-LAPW method, Berri et al. [10] predicted a ferromagnetic half-metallic behavior of ZrFeTiAl, ZrFeTiSi, ZrFeTiGe, and ZrNiTiAl alloys with total magnetic moments of 1, 2, 2, and 3 μB/f.u., respectively, and large gap values between 0.56 and 0.92 eV. Furthermore, Wei et al. predicted half-metallic ferromagnetic structure of NiCoCrAl, NiCoCrGa, and NiFeCrGa alloys [11]. Moreover, FeMnScZ (Z = Al, Ga, and In) alloys were predicted to be half-metallic ferromagnets with total magnetic moments of 3 μB/f.u. [12]. Another series of CoFeMnZ (Z = Si, As, and Sb) quaternary Heusler alloys were predicted to have ferromagnetic half-metallic behavior with high magnetic moment and Curie temperature [13]. Recently, Mohamedi et al. [14] reported a theoretical study of the elastic, electronic, and thermal properties of CoMnCrZ (Z = Al, As, Si, and Ge) compound using FP-LAPW method. The authors pointed out that the type 1 structure with the FM configuration is energetically more favorable than types 2 and 3 structures. It has been found that the CoMnCrGe, CoMnCrAl, CoMnCrSi, and CoMnCrAs quaternary Heusler compounds are half-metallic ferromagnets with half-metallic gaps of 0.03, 0.19, 0.34, and 0.50 eV, respectively.
Motivated by the promising properties of the quaternary Heusler compounds specially those containing Co, V, and Ti elements, we performed computational investigation by first-principle calculations to study the structural, mechanical, electronic, and magnetic properties of CoVTiX (X = As, Si) alloys. To our knowledge, there is no comprehensive theoretical or experimental study on these compounds. The rest of the paper is organized as follows: Section 2 outlines the computational methods used in our study. The results discussed in this paper are obtained using density functional theory (DFT) and are presented in Section 3, and Section 4 summarizes our main results.
2 Computational Details
First-principle calculations were performed using the FP-LAPW method as implemented in the Wien2K [15] code to study the structural, mechanical, and magnetic properties of CoVTiX (X = As, Si) compounds. The electron-electron interaction was treated within the generalized gradient approximation (GGA) by Perdew, Burke, and Ernzerhof (PBE) exchange correlation potential [16] in the case of the structural and mechanical properties. However, the modified Becke-Johnson potential (mBJ) [17] was adopted to obtain the electronic structure. The expansion of the valence wave functions inside the non-overlapping muffin-tin spheres were confined to lmax = 10, while the charge density was Fourier expanded up to Gmax = 14. In order to achieve energy convergence, the wave functions in the interstitial region were expanded in plane waves with a cutoff of Kmax = 8/RMT where RMT denotes the smallest atomic sphere radius and Kmax gives the magnitude of the largest K vector in the plane-wave expansion. The RMT are taken to be in the range 1.8–2.15 atomic units (a.u.) for Co, V, Ti, As, and Si atoms for all types. Brillouin zone (BZ) integrations within the self-consistency cycles were performed via the tetrahedron method [18, 19] using 47k points for CoVTiX (X = As, Si) in their irreducible BZ (IBZ). The energy convergence was obtained when the difference is less than 0.1 mRy.
3 Results and Discussions
In this section, we present the structural, mechanical, and electronic properties of CoVTiX (X = As, Si) alloys.
3.1 Structural Properties
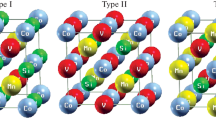
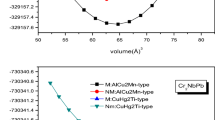
The quaternary Heusler alloys have XX′YZ chemical structure with 1:1:1:1 stoichiometry, where X, X′, and Y are transition metals and Z is a main group element. These compounds crystallize in the LiMgPdSn crystal structure with a space group \(F\overline {4} 3m\). In general, in LiMgPdSn-type quaternary Heusler alloys can have three different types as shown in Fig. 1. These types are as follows: (i) type 1: X (0, 0, 0), X′ (1/4, 1/4, 1/4), Y (1/2, 1/2, 1/2), and Z (3/4, 3/4, 3/4), (ii) type 2: X (0, 0, 0), X′ (1/2, 1/2, 1/2), Y (1/4, 1/4, 1/4), and Z (3/4, 3/4, 3/4), and (iii) type 3: X (0, 0, 0), X′ (1/4, 1/4, 1/4), Y (1/2, 1/2, 1/2), and Z (3/4, 3/4, 3/4). In order to obtain the ground state structure of each alloy and find the favorable magnetic state, we performed total energy calculations as a function of volume for the three different possible site occupations for non-magnetic (NM), ferromagnetic (FM), and antiferromagnetic (AFM) states. The structures of all quaternary Heusler alloys were optimized by calculating the total energy as a function of volume, which was followed by fitting the data with Murnaghan equation of state [20]. The site occupation preference is obtained by comparing the total energy versus volume of the three types of CoVTiX (X = As, Si) compounds as shown in Fig. 2. From this figure, one can see that CoVTiAs and CoVTiSi prefer to crystallize in the type 2 structure. Our calculations show that the energy difference of the two types (2 and 3) is only about ΔE = 10− 4 Ry. The most stable magnetic structure is found to be the FM structure as compared with the AFM and NM states for CoVTiX (X = As, Si) compounds in all types (1, 2, and 3), see Fig. 3. In Fig. 3a, b, type 2 of both compounds CoVTiAs and CoVTiSi has the lowest energy curve. Therefore, it is clear that the most stable phase at ambient pressure is type 2 structure. Similar compounds such as CoMnCrZ (Z = Al, As, Si, and Ge) alloys were found to prefer type 1 (FM) structure [14]. In Table 1, we report our calculated equilibrium lattice constants, along with bulk modulus, pressure derivatives (B′), and the total energy in their different types compared with previous works [21]. From this table, one can see that the lattice constant decreases, whereas the bulk modulus increases when the Z element decreases.

Crystal structure of CoVTiX (X = As, Si) at different types

Variation of total energies as a function of volume of unit cell using GGA for CoVTiX (X = As, Si) at different types

Variation of total energies as a function of volume of unit cell using GGA for CoVTiX (X = As, Si) in the three magnetic states FM, AFM, and NM
3.2 Elastic Properties and Mechanical Stability
The knowledge of elastic constants of a solid provides valuable information on their dynamical and mechanical properties. The elastic constants Cij for these quaternary Heusler alloys are calculated to determine their mechanical behavior. It is well known that a cubic crystal has only three independent elastic constants, C11, C12, and C44. There are three conditions that should be satisfied in order to have mechanical stability in a cubic structure [22]: C11 + 2C12 > 0; C11 − C12 > 0; and C44 > 0. Accordingly, we found that the compounds are elastically stable in the cubic structure except for the case of CoVTiAs in the type 1 structure, see Table 2.
The shear (CS), Voigt (CV), and Reuss (CR) moduli can be expressed as follows [23,24,25]:
The Voigt-Reuss-Hill approximation [26, 27] could be approximated by the arithmetic mean of the two well-known limits for mono-crystals according to Voigt [28] and Reuss [29] and is given by the following expression [27, 30]:
For the cubic system, the modulus of rigidity B can be expressed as a linear combination of the two elastic constants C11 and C12 given by [25]:
The elastic constants are found to increase when As is replaced by Si in type 2. The elastic constant values of C44 for all compounds are found to be far less than the values of C11, which suggests that these quaternary Heusler alloys show weaker resistance to bulk strains. To the best of our knowledge, there are no experimental or theoretical results for the elastic constants of these compounds reported yet; hence, our results serve as a prediction for future investigations. The calculated values of the bulk modulus (B), Young’s modulus (E), and Poisson’s ratio (𝜗) of the title materials using VRH approximations in GGA are listed in Table 2. The higher calculated bulk moduli for CoVTiX (X = As, Si) in type 2 confirm the stability of this structure. It is noted that the higher bulk and Young’s moduli suggest a strong incompressibility for these compounds. The Young’s modulus is the property of a material, which is used to characterize stiffness. The higher the value of E, the stiffer is the material. Young’s modulus is found to increase when replacing As by Si, which means that CoVTiAs is stiffer than CoVTiSi. In addition, Poisson’s ratio for CoVTiX (X = As, Si) is found to fall in this limit, which indicates that the interatomic forces in this compounds are central forces. This is ascribed to the fact that Poisson’s ratio provides more information about the characteristics of the bonding forces than any of the other elastic constants [31]. It has been proven that σ = 0.25 is the lower limit for central force and 0.5 is the upper limit [31]. Therefore, the high σ values of CoVTiAs (σ = 0.4) and CoVTiSi (σ = 0.3) indicate that the interatomic forces in the title compound are central. Moreover, the Debye temperature of CoVTiAs is found to be lower than that of CoVTiSi.
The anisotropy factors (A) of the alloys can be expressed as follows:
It is found that A is higher than 1 in all types of both alloys except for CoVTiAs in type 1, which means that the crystals are harder in the < 111> direction. To complete the study of the mechanical properties, we obtained a three-dimensional (3D) surface, representing the dependence of the Young’s modulus on crystallographic directions, which is an effective method to visualize the elastic anisotropy of a material. The 3D closed surfaces that represent the dependence of E on the crystallographic directions of a cubic crystal is defined as follows [23]:
where \(S_{ij} = C_{ij}^{-1}\)
l1 = sin𝜃 cosφ, l2 = sin𝜃 sinφ, and l3 = cos𝜃 are the directional cosines with respect to the x, y, and z axes, respectively. The obtained 3D closed surfaces of the Young’s modulus of type 2 CoVTiAs and CoVTiSi are depicted in Figs. 4 and 5, respectively. It is found that the shapes of these surfaces are clearly different from the spherical shape, which indicates the presence of a strong elastic anisotropy in the studied quaternary Heusler alloys. We also plotted the cross-sections of these surfaces in different planes. From the 2D plane projections, one can see that the Young’s modulus at different planes has a large anisotropic character.

3D representation of directional dependence of Young’s modulus and cross-section in some reticular planes of 3D representation of CoVTiAs type 2

3D representation of directional dependence of Young’s modulus and cross-section in some reticular planes of 3D representation of CoVTiSi type 2
3.3 Electronic and Magnetic Properties
In this subsection, we present the electronic and magnetic properties of these quaternary Heusler alloys using GGA and mBJ functionals. The band structures of CoVTiX (X = As, Si) alloys were calculated using the theoretical equilibrium lattice constants along high symmetry directions of the first Brillouin zone, see Fig. 6. Both CoVTiAs and CoVTiSi are found to exhibit a metallic behavior in the case of type 1 structure using GGA and mBJ approximations. For the case of type 2 CoVTiAs, GGA calculations predict a metallic behavior in the spin-up and spin-down channels, see Fig. 6a. However, using the GGA-mBJ approximation, the minority spin remains metallic, whereas the majority spin exhibits a metallic behavior due to the downward and upward shifts of the valence band maximum (VBM) and conduction band minimum (CBM), respectively. The majority spin band of the type 2 CoVTiAs alloy is found to be 0.45 eV, which extends from the Γ high symmetry point at the VBM to the X high symmetry point at the CBM that leads to a 100% spin polarization at the Fermi level. Similarly, CoVTiAs and CoVTiSi alloys of type 3 show half metallicity due to the semiconducting and metallic characters of the majority and minority spin channels, respectively, in agreement with CoMnCrZ (Z = Al, Si, Ge, and As) alloys [14]. The results of GGA-mBJ approximation are believed to present good predictions for the band structures. It has been found that the band gaps of 15 out of 27 semiconductors are in agreement with experiment within a 5% error or less for both GW and the mBJ potentials [32]. However, the computations using GGA-mBJ functional is less time consuming with reasonable results as compared with GW. Previous electronic structure calculations of Fe2VAl alloy, using GGA-mBJ functional, predicted a band gap of 0.22 eV [33], which is in a good agreement with the experimental values in the range of 0.1–0.2 eV [34]. A half-metallic behavior is also predicted for the case of CoVTiSi, where the minority band structure exhibits a metallic structure, whereas the majority spin channel is semiconducting. It is obvious that the band gap decreases when replacing As by Si. The band gap values are listed in Table 3.To confirm the nature of band structure of the studied compounds, the total and partial densities of states are depicted in Fig. 7. From this figure, one can see that the region between − 13 and − 11.5 eV of CoVTiAs shows the contribution of 4s orbitals of As atom for both majority and minority spin channels and two bands between − 6 and − 3 belong to p states with small contributions from the Cod states.The region extending from − 4 eV to the Fermi energy is mainly formed by Co, V, and Tid states. The d states of Co, V, and Ti transition metals are found to be the main contribution of the conduction band. For CoVTiSi compound, in both the majority and minority spin channels, significant contributions to the total density of states in the energy range between − 9 and − 7 eV, come from s states of Si atom. The second region of the valence band from − 6 to − 3 eV contains the small contributions of Si p orbitals and Co d orbitals. The remaining region between − 3 and 5 eV shows a strong hybridization of Co, V, and Ti 3d orbitals. The density of states also confirms that these quaternary Heusler alloys are half-metallic ferromagnets using mBJ functional, which suggests these compounds as candidate materials for future spintronic applications. In Table 4, we present the calculated total and partial magnetic moments per formula unit for the studied quaternary Heusler compounds. The total moments of type 2 CoVTiAs and CoVTiSi alloys are found to be 1 and 2 μB, respectively. The main contribution of the total magnetic moment comes from Co and V local magnetic moments, which are found to be ferromagnetically coupled. However, Ti and As, and Si exhibit very small local magnetic moments. The magnetic moment is defined as the difference between the integral of the spin-up and spin-down densities of states. This explains the higher total and local magnetic moments in the case of CoVTiSi than those of CoVTiAs, which is attributed to the shift of the d states in the valence band of the minority spin channel to higher energy levels in the case of CoVTiSi as compared with CoVTiAs, see Fig. 7. To the best of our knowledge, there are neither experimental nor theoretical magnetic moments obtained for these quaternary Heusler compounds in the literature.


Band structures for CoVTiX (X = As, Si) in type 2 structure calculated using GGA and GGA-mBJ approximations


Partial and total density (DOS) CoVTiX (X = As, Si) with GGA and GGA-mBJ approximations in type 2 structure
4 Conclusions
The structural, electronic, magnetic, and mechanical properties of CoVTiX (X = As, Si) quaternary Heusler alloys were investigated using DFT calculations. Three possible different types were investigated for these alloys, where the type 2 ferromagnetic structure was found to be energetically more favorable than types 1 and 3. The mechanical properties reveal that the studied compounds are mechanically stable in their energetically stable phase. This study shows that these quaternary Heusler compounds possess a gap in the majority spin channel with a half-metallic ferromagnetic behavior, which is suitable for spintronic applications. The results in this work appear promising for future experimental investigations.
References
Galanakis, I., Dederichs, G.A., Papanikolaou, N.: J. Phys. D 39, 765 (2006)
Xu, D., Liu, G., Fecher, G.H., Felser, C., Li, Y., Liu, H.: J. Appl. Phys. 105, 07E901 (2009)
Xu, G.Z., Liu, E.K., Du, Y., Li, G.J., Liu, G.D., Wang, W.H., Wu, G.H.: Europhys. Lett. 102, 17007 (2013)
Galanakis, I.: J. Phys.: Condens. Matter 16, 3089 (2004)
Balke, B., Fecher, G.H., Kandpal, H.C., Felser, C.: Phy. Rev B 74, 104405 (2006)
Alijani, V., Winterlik, J., Fecher, G., Naghavi, S., Felser, C.: Phys. Rev. B 83, 184428 (2011)
Gao, G.Y., Hu, L., Yao, K.L., Luo, B., Liu, N.: J. Alloy. Compd. 551, 539 (2013)
Nehra, J., Sudheesh, V.D., Lakshmi, N., Venugopalan, K.: Phys. Status Solidi RRL 7, 289 (2013)
Bainsla, L., Suresh, K.G., Nigam, A.K., Manivel Raja, M., Varaprasad, B.S.D.Ch.S., Takahashi, Y.K., Hono, K.: J. App. Phys. 116, 203902 (2014)
Berri, S., Ibrir, M., Maouche, D., Attallah, M.: J. Magn. Magn. Mater. 371, 106 (2014)
Wei, X.P., Zhang, Y.L., Chu, Y.D., Sun, X.W., Sun, T., Guo, P., Deng, J.B.: J. Phys. Chem. Solids 82, 28 (2015)
Gao, Y.C., Gao, X.: AIP Adv. 5, 057157 (2015)
Elahmar, M.H., Rached, H., Rached, D., Khenata, R., Murtaza, G., BinOmran, S., Ahmed, W.K.: J. Magn. Magn. Mater. 393, 165 (2015)
Mohamedi, M.W., Chahed, A., Amar, A., Rozale, H., Lakdja, A., Benhelal, O., Sayede, A.: Eur. Phys. J. B 89, 267 (2016)
Blaha, P., Schwarz, K., Madsen, G.K.H., Kvasnicka, D., Luitz, J.: WIEN2K, Karlheinz Schwarz, Techn. Universitat, Wien, Austria. ISBN 3-9501031-1-1-2 (2001)
Perdew, J.P., Burke, S., Ernzerhof, M.: Phys. Rev. Lett. 77, 3865 (1996)
Tran, F., Blaha, P.: Phys. Rev. Lett. 102, 226401 (2009)
Jepsen, O., Andersen, O.K.: Sol. Stat. Commun. 9, 1763 (1971)
Wilson, J.A., Yoffe, A.D.: Phys. Adv. 18, 193 (1969)
Murnaghan, F.D.: Proc. Nat. Acad. Sci. USA 30, 244 (1944)
Ozdogan, K., Sasiogolu, E., Galanakis, I.: J. Appl. Phys. 113, 193903 (2013)
Born, M., Huang, K.: Dynamical Theory of Crystal Lattices. Clarendon, Oxford (1956)
Nye, J.F.: Properties of Crystals. Oxford University Press, New York (1985)
Adachi, S.: Physical Properties of III-V Semiconductor Compounds. Wiley, New York (1992)
Adachi, S.: Properties of Group-IV, III-V and II-VI Semiconductors. Wiley, New York (2005)
Grimvall, G.: Thermo Physical Properties of Materials. North-Holland, Amsterdam (1999)
Zhao, H., Chang, A., Wang, Y.: Physica B 404, 2192 (2009)
Voight, W.: Johnson Reprint Corp (1928)
Reuss, A., Angew, Z.: Math. Mech. 9, 49 (1929)
Bing, L., Feng, L.R., Yong, Y., Dong, Y.X.: Chin. Phys. B 19, 076201 (2010)
Ravindran, P., Fast, L., Korzhavyi, P.A., Johansson, B.: J. Appl. Phys. 84, 4891 (1998)
Camargo-Martínez, J.A., Baquero, R.: Rev. Mex. Fis. 59, 453 (2013)
Shastri, S.S., Pandey, S.K.: Comput. Mater. Sci. 143, 316 (2018)
Okamura, H., Kawahara, J., Nanba, T., Kimura, S., Soda, K., Mizutani, U., Nishino, Y., Kato, M., Shimoyama, I., Miura, H., Fukui, K., Nakagawa, K., Nakagawa, H., Kinoshita, T.: Phys. Rev. Lett. 84, 3674 (2000)
Funding
This work is supported by the Algerian University research project (CNEPRU) under No. B00L02UN280120140051
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Mokhtari, D., Baaziz, H., Guendouz, D. et al. Theoretical Investigation of Structural, Electronic, Magnetic, and Mechanical Properties of Quaternary Heusler Alloys CoVTiX (X = As, Si). J Supercond Nov Magn 31, 3625–3636 (2018). https://doi.org/10.1007/s10948-018-4614-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10948-018-4614-y