
Abstract
The geometrical structure of CdSe was optimized by using the ultrasoft pseudopotential method of a total energy plane wave based on density functional theory. The band structure, density of states, and optical properties were calculated and discussed in detail. The Mn-doped CdSe is found to be a half-metallic ferromagnet with 100% carrier spin polarization at the Fermi level. At a Mn concentration of 12.5%, the calculated total energy of the spin-polarized state is 614 meV lower than that of the nonspin-polarized state. The net magnetic moment of 5 μ B is found per supercell for 12.5% Mn-doped CdSe. The estimated Curie temperature of 748.6 K for Mn-doped CdSe is above room temperature. The ferromagnetic ground state in Mn-doped CdSe can be explained in terms of the p − d hybridization mechanism. These results suggest that Mn-doped CdSe may present a promising dilute magnetic semiconductor, and may have potential applications in the field of spintronics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
In recent years, dilute magnetic semiconductors (DMSs), fabricated by doping conventional semiconductors with a transition metal or rare earth element, have attracted more and more research interests because of their promising application in spintronic devices [1,2,3]. By introducing transition metal dopants or rare earth elements, quantum size effects and atomic band transitions are combined, resulting in a novel class of materials with new electronic, optical, and magnetic properties [4]. For practical applications, it is required to fabricate a high Curie temperature (T C) ferromagnet with a homogeneous distribution of the magnetic dopants. Since the original reports of ferromagnetism in Mn-doped InAs and GaAs above 100 K [5, 6], II–VI based semiconductors have been intensively studied for they are typical semiconductors in which the carrier control technique is well established and they display ferromagnetism when doped with most of the transition-metal elements. Extensive work has been reported on various DMS systems such as CdS [7,8,9], ZnO [10,11,12], CdTe [13], ZnS [14, 15], ZnSe [16, 17], and CdSe [18, 19]. Among all these widely studied DMSs and II–VI group DMSs, one of particular interest is CdSe, because it is a versatile material for spintronics and magneto-optical device applications [20, 21]. Ahmadian et al. [22] have investigated the structural, electronic and magnetic properties of V-doped CdSe by performing the first-principles calculations and confirmed there exhibit a half-metallic characteristic. Oluwafemi et al. [23] have successfully synthesized Mn-doped wurtzite CdSe, and the nanoparticles obtained by them show quantum confinement in optical properties. The half-metallic ferromagnetic and optical properties of Fe-doped and Co-doped CdSe have been investigated in a recent work of one of the author’s [24]. The interest in the Mn-doped CdSe is based on their outstanding magneto-optical properties caused by a strong s,p − d exchange interaction [25,26,27] between electron/hole band states and Mn 2+ 3d electron states, but the properties of it have received little attention. The interaction between Mn 3d electron and the electronic states of the host crystal is of special interest [28].
In this paper, the geometrical structure of Mn-doped CdSe was optimized by using the ultrasoft pseudopotential method of total energy plane-wave based on density functional theory. The density of states, band structure, and magnetic and optical properties are calculated and discussed in detail. The layout of this paper is given as follows: The detailed calculations and employed methods are given in Section 2. Results and discussions of the structural, optical, electronic, and magnetic properties will be presented in Section 3. Conclusions are given in Section 4.
2 Model and Calculation Methods
The first-principles calculations were implemented in the CASTEP code [29] based on the spin-polarized density functional theory (SP-DFT) method [30]. Ultrasoft pseudopotentials [31] are chosen to represent the interactions between the ionic core and valence electrons. The valence electron configurations considered in this study included Cd: 4 d 105s 2, Se: 4 s 24p 4, and Mn: 3 d 54s 2. The Perdew and Wang parametrization of GGA and PBESOL are adopted for the exchange-correlation potential [32,33,34,35]. The electron wave function is expanded in plane waves up to a cutoff energy of 500 eV. A gamma-centered 3 × 3 × 2 k mesh is used to sample the irreducible Brillouin zone of bulk CdSe. These parameters ensure a convergence in total energy. The maximum force on each atom is less than 0.01 eV/Å. The maximum energy on a single atom is less than 5 × 10−6 eV/atom. The maximum stress of the crystal is less than 0.02 GPa. The maximum displacement of the atom is less than 5 × 10 −4 Å. In the structure optimization, the Brodyden-Fletcher-Goldfarb-Shanno (BFGS) algorithm [36] is used.
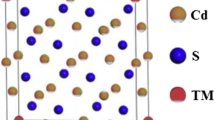
CdSe possesses a hexagonal wurtzite (WZ) structure at ambient condition. The experimental lattice constants a and c of pure wurtzite CdSe are a = 4.299 Å and c = 7.010 Å [37]. The supercell employed contains 32 atoms which correspond to a 2 × 2 × 2 supercell of CdSe. Two Cd atoms are replaced by Mn atoms from the 2 × 2 × 2 supercell, as is illustrated in Fig. 1. The doped structures were then optimized with respect to both the lattice constants and the atomic positions.

Crystal structure of Mn-doped CdSe
3 Results and Discussion
3.1 Structural Properties
The calculated lattice constants a and c of pure wurtzite CdSe are a = 4.307 Å and c = 7.012 Å, which are in good agreement with the experimental values [37]. The result of c/a = 1.628 is consistent with previous experimental and theory work [38,39,40]. When two Cd atoms in the 2 × 2 × 2 CdSe supercell are replaced by Mn, the calculated lattice constants are a = 4.285 Å and c = 6.971 Å. The lattice constants can be calculated from the parameter d [25] as follows:
where d is the mean cation-cation distance; d = 4.296 − 0.123x for Cd1−xMn x Se (x = 0.125). The calculated lattice parameters of Cd1−xMn x Se compounds are a = 4.281 Å and c = 6.989 Å which are in good agreement with the data of a = 4.285 Å and c = 6.971 Å. There is some decrease in the lattice constants compared with pure wurtzite CdSe after structural optimization, which can be attributed to the fact that the ionic radius of Cd is larger than that of Mn.
The mixing energy for Cd1−xMn x Se (x = 0.125) was calculated as follows:
where \(E_{\text {tot}}^{\text {Cd}_{1-x} \text {Mn}_{x} \text {Se}} \), \(E_{\text {tot}}^{\text {CdSe}} \), and \(E_{\text {tot}}^{\text {MnSe}} \) are total energy of zinc blende Cd1−xMn x Se compounds, the pure zinc blende CdSe, and pure MnSe, respectively. The calculated mixing energy of Cd1−xMn x Se (x = 0.125) compounds was 0.331 eV which illustrates that the zinc blende Cd1−xMn x Se (x = 0.125) compounds are unstable.
3.2 Electronic and Magnetic Properties
In order to understand the ferromagnetic properties from the microscopic point of view, we inspect the band structures of both pure CdSe and doped CdSe, as shown in Figs. 2a and 3 Both the band structure and partial density of states (PDOS) of pure CdSe are calculated at their equilibrium lattice constants within GGA-PBESOL schemes, and they are presented in Fig. 2. From Fig. 2a, we can see the pure CdSe is a direct band gap semiconductor and its band gap value is 1.74 eV [41]. The states in the energy range from −4.4 to 0 eV are attributed to Se-4p4s and Cd-5s states, and the states in the energy range from −12.6 to −7.1 eV mainly originate from Cd-4d and Se-4 p4s states with small contributions from Cd-5s states. Especially, there is a strong localized state at −7.5 eV which is attributed to Cd-4d. The conduction band mainly consists of Se-4 p4s and Cd-5s states. Figure 3 shows the band structure of the majority spin and the minority spin of CdSe doped with 12.5% of Mn. The valence band and conduction band meet at the Fermi level in minority spin states, while in majority spin states, there is an energy gap confirming the half-metallic (HM) characteristic of Cd1−xMn x Se (x = 12.5%).

The band structure (a) and partial density of states (b) of pure supercell CdSe

Band structures of the majority spin (a) and the minority spin (b) of CdSe with 12.5% of Mn. The Fermi level is set to zero
To analyze the detailed electronic structure and the origin of the impurity energy levels of the Mn-doped CdSe, the total spin density of states (TDOS) and PDOS of these systems were all calculated, as depicted in Fig. 4. We mainly discuss the DOS near the Fermi level because magnetic properties are decided by this. From Fig. 4a, it can be found that there is a large exchange splitting between the majority and minority spin states around the Fermi level. The majority spin electrons have a metallic character while there is a band gap at the Fermi level in the minority spin states. On the other hand, we calculate the integration of the majority spin and the minority spin of the total spin density of states that is below the Fermi level. It turns out that the electrons of the majority spin are more than those of the minority spin. Therefore, the HM characteristic of Cd1−xMn x Se (x = 12.5%) is confirmed. The calculated total energy for the spin-polarized state is 614 meV lower than that of the nonspin-polarized state, which indicates that the Mn-doped CdSe compounds prefer the ferromagnetic ground state. The calculated total magnetic moment is 5 μ B per supercell in the ferromagnetic (FM) state. The lattice constants, bond length, energy gap, and magnetic moment of Cd1−xMn x Se (x = 6.25%) were investigated for comparison. The results are listed in Table 1.

The total spin density of states (a), partial density of states (b), Mn-3d spin density of states (c), Se-4p spin density of states (d), and total density of states (e). The Fermi level is set to zero
The PDOS of Cd1−xMn x Se (x = 12.5%) is depicted in Fig. 4b. The lower valence band is mainly attributed to Cd-4d and Se-4s states, which is the same as that of pure CdSe. The conduction band mainly consists of Mn-3d states, and the other valence band mainly consists of Mn-3d and Se-4p states. We compared Fig. 4c, d and found that the peaks at −5, −3.8, and −1.7 eV of Mn-3d overlap that of 4p of the Se atom in the majority spin. Especially at −5 and −3.8 eV, there is strong hybridization between Mn and Se atoms. We also calculate the integration of the majority spin and the minority spin of Mn-3d and Se-4p below the Fermi level. And the electrons of the majority spin are more than those of the minority spin. This induces finite magnetizat ion on Mn-3d as well as Se-4p. Using p − d hybridization, we can explain the exchange interactions in Mn-doped CdSe DMS. In the framework of the band structure, p − d hybridization essentially means the following: the dopant hybridizes strongly with its neighboring anions of the host semiconductors, then the neighboring anions are spin polarized with large magnetization and couple ferromagnetically or antiferromagnetically with the dopant, which results in an energy gain. In the present case, the conditions mentioned above are fulfilled. Therefore, the mechanism p − d hybridization dominates the ferromagnetism of the present system. Figure 4e shows TDOS after doping. We can see the Fermi level moves into the upper valence band. A few electron holes appear above the Fermi level. It indicates that Mn-doped CdSe belongs to p-type dopant.
The Curie temperature (T C) can be estimated with the classical Heisenberg model in the mean-field theory [42]:
where x is the concentration of the doping atoms and k B is Boltzmann’s constant. By (4), the T C of Mn-doped CdSe is obtained. It is about T C = 748.6 K and higher than room temperature. The result illustrates that Mn-doped CdSe is more promising spintronics material.
3.3 Optical Properties
The optical properties are important for compounds, since they can find potential applications in photoelectron devices and the semiconductor industry. The optical properties are studied by dielectric function ε = ε 1 + i ε 2; the imaginary part of the dielectric constant (ε 2) can be calculated theoretically based on DFT (See (5) as follows), and then, by applying the Kramers-Krönig relation, we can evaluate the real part (ε 1) at the same time. The peaks appearing in the ε 2 part of the dielectric function are directly related to different intra-band or inter-band transitions in the first irreducible Brillouin zone.
Figure 5a, b illustrates the real and imaginary parts of the dielectric function of the pure CdSe and Mn-doped CdSe in the range from 0 to 20 eV. The real part of the dielectric function governs the propagation behavior of the electromagnetic field in a material. The static dielectric constants ε 1 (0) obtained for pure CdSe, Cd1−xMn x Se (x = 6.25%), and Cd1−xMn x Se (x = 12.5%) were 9.313, 7.878, and 15.222, respectively. The imaginary part of the dielectric function represents the light absorption in the crystal. From Fig. 5b, the peaks of the spectra are at 1.4, 4.4, 6.3, and 10.7 eV for pure CdSe; 1.6, 5.7, and 15.5 eV for Cd1−xMn x Se (x = 6.25%); and 0.7, 5.5, and 15.4 eV for Cd1−xMn x Se (x = 12.5%), respectively.

Real (a) and imaginary (b) parts of the dielectric function of pure CdSe (black line), Cd1−xMn x Se (x = 6.25%) (red line), and Cd1−xMn x Se (x = 12.5%) (green line)
The absorption coefficient is calculated by [43]
where λ, w, ε 1, and ε 2 represent the wavelength, frequency of the incident light, and the real and the imaginary parts of the dielectric constant. The corresponding absorption spectra and reflectivity spectra of the pure CdSe, Cd1−xMn x Se (x = 6.25%), and Cd1−xMn x Se (x = 12.5%) are presented in Fig. 6. For the pure CdSe, there are five peaks of absorption spectra. The energy of these peaks is 7.8, 11.0, 13.8, 17.5, and 19.0 eV. There is some difference between the energy of absorption peaks and the energy of dielectric peaks. It is believed that the difference is caused by considering the relaxation effect of electronic transition when calculating the absorption coefficient. With the increasing of the electromagnetic energy, the absorption energy of CdSe increases rapidly until 7.8 eV, then decreases gradually. After doping, the energy of the absorption peaks is 7.5 and 15.8 eV for Cd1−xMn x Se (x = 6.25%) and 7.2 and 15.6 eV for Cd1−xMn x Se (x = 12.5%), respectively. From Fig. 6b, we can see that the reflectivity strengthened between 7.5 and 12.5 eV after doping. That may be the reason that the doped systems have weak absorption than pure CdSe in the UV-light range and visible region.

Absorption (a) and reflectivity (b) spectra of pure CdSe (black line), Cd1−xMn x Se (x = 6.25%) (red line), and Cd1−xMn x Se (x = 12.5%) (green line)
4 Conclusions
In summary, our accurate first-principles simulations predict Mn-doped CdSe to be a half-metallic dilute magnetic semiconductor. Two kinds of doping concentration of Mn have been studied using first-principles method based on DFT. At a Mn concentration of 12.5%, the calculated total energy for the spin-polarized state is 614 meV lower than that of the nonspin-polarized state. The net magnetic moments of 2.47 and 5 μ B are found per supercell for 6.25% and 12.5% Mn-doped CdSe, respectively. The ferromagnetism emerges from the hybridization of the Mn-3d and Se-4p bands. We also briefly discuss the complex dielectric function, absorption spectrum, and reflectivity spectrum. With the 100% spin polarization of the carriers, we expect Mn-doped CdSe to be a useful DMS, both in theoretical studies and applications of the ferromagnetism in DMSs.
References
Ohno, H.: Science 281, 951 (1998)
Matsumoto, Y., Murakami, M., Shono, T., Hasegawa, T., Fukumura, T., Kawasaki, M., Ahmet, P., Chikyow, T., Koshihara, S., Koinumaal, H.: Science 291, 854 (2001)
Das, G.P., Pao, B.K., Jena, P.: Phys. Rev. B 69, 214422 (2004)
Norris, D.J., Efros, A.L., Erwin, S.C.: Science 319, 1776 (2008)
Munekata, H., Ohno, H., Von Molnar, S., Segmüller, A., Chang, L.L., Esaki, L.: Phys. Rev. Lett. 63, 1849 (1989)
Ohno, H., Shen, A., Matsukura, F., Oiwa, A., Endo, A., Katsumoto, S., Lye, Y.: Appl. Phys. Lett. 69, 363 (1996)
Elango, M., Gopalakrishnan, K., Vairam, S., Thamilselvan, M.: J. Alloys Compd. 538, 48 (2012)
Ghosh, A., Paul, S., Gopal, R.K., Raj, S.: Phys. Status Solidi B 252, 1355 (2015)
Ghosh, A., Paul, S., Raj, S.: J. Magn. Magn. Mater. 405, 238 (2016)
Liu, C., Yun, F., Morkoc, H.: J. Mater. Sci.: Mater. Electron. 16, 555 (2005)
Yuan, H., Zhang, L., Xu, M., Du, X.S.: J. Alloys Compd. 651, 571 (2015)
Varadhaseshan, R., Meenakshi, S.S., Ganesan, V., Banerjee, A., Lalla, N.P.: Optik 127, 1499 (2016)
Ebrahim, S.h., Ramadan, W., Ali, M.: J. Mater. Sci: Mater. Electron 27, 3826 (2016)
Chu, D.W., Zeng, Y.P., Jiang, D.L.: Solid State Commun. 143, 308 (2007)
Zhang, C.W., Yan, S.S.: J. Appl. Phys. 107, 043913 (2010)
Jonker, B.T., Chou, W.C., Petrou, A., Warnock, J.: J. Vac. Sci. Technol. A 10, 1458 (1992)
Chin, P.T.K., Stouwdam, J.W., Janssen, R.A.J.: Nano Lett. 9, 745 (2009)
Kumar, S., Kumar, S., Verma, N.K., Chakarvarti, S.K.: J. Mater. Sci: Mater. Electron 22, 901 (2011)
Li, Z., Du, A.J., Sun, Q., Aljada, M., Cheng, L.N., Riley, M.J., Zhu, Z.H., Cheng, Z.X., Wang, X.L., Hall, J., Krausz, E., Qiao, S.Z., Smith, S.C., Lu, G.Q.: Chem. Commun. 47, 11894 (2011)
Singh, J., Verma, N.K.: J. Supercond. Nov. Magn. 25, 2425 (2012)
Singh, S.B., Limaye, M.V., Date, S.K., Gokhale, S., Kulkarni, S.K.: Phys. Rev. B 80, 235421 (2009)
Ahmadian, F., Makaremi, N.: Solid State Commu. 152, 1660 (2012)
Oluwafemi, O.S., Revaprasadu, N., Adeyemi, O.O.: Mater. Lett. 64, 1513 (2010)
Tian, J.H., Song, T., Sun, X.W., Wang, T., Jiang, G.: J. Supercond. Nov. Magn. 30, 521 (2017)
Furdyna, J.K.: J. Appl. Phys. 64, 29 (1988)
Nikitin, P.I., Savchuk, A.I.: Sov. Phys. Usp. 33, 974 (1990)
Norberg, N.S., Gamelin, D.R.: J. Appl. Phys. 99, 08M104 (2005)
Weidemann, R., Gumlich, H.E., Kupsch, M., Middelmann, H.U.: Phys. Rev. B 45, 1172 (1992)
Segall, M.D., Philip, J.D., Lindan, M.J., Probert, C., Pickard, J., Hasnip, P.J., Clark, S.J., Payne, M.C.: J. Phys.: Condens. Matter 14, 2717 (2002)
Dudarev, S.L., Botton, G.A., Savrasov, S.Y., Humphreys, C.J., Sutton, A.P.: Phys. Rev. B 57, 1505 (1998)
Vanderbilt, D.: Phys. Rev. B 41, 7892 (1990)
Perdew, J.P., Chevary, J.A., Vosko, S.H., Jackson, K.A., Pederson, M.R., Singh, D.J., Fiolhais, C.: Phys. Rev. B 46, 6671 (1992)
Perdew, J.P., Ruzsinszky, A., Csonka, G.I., Vydrov, O.A., Scuseria, G.E., Constantin, L.A., Zhou, X.L., Burke, K.: Phys. Rev. Lett. 101, 239702 (2008)
Mattsson, A.E., Armiento, R., Mattsson, T.R.: Phys. Rev. Lett. 101, 239701 (2008)
Pierre, M.D., Orlando, R., Maschio, L., Doll, K., Ugliengo, P., Dovesi, R.: J. Comput. Chem. 32, 1775 (2011)
Fische, T.H., Almlof, J.: J. Phys. Chem. 96, 9768 (1992)
Reeber, R.R.: J. Mat. Sci. 11, 590 (1976)
Sarasamak, K., Limpijumnong, S., Lambrecht, W.R.L.: Phys. Rev. B 82, 035201 (2010)
Hotje, U., Rose, C., Binnewies, M.: Solid State Sci. 5, 1259 (2003)
Sowa, H.: Solid State Sci. 7, 1384 (2005)
Kittel, C.: Introduction to Solid State Physics, 8th edn. Wiley, New York (2005)
Perdew, J.P., Ruzsinszky, A., Csonka, G.I., Vydrow, O.A., Scuseria, G.E., Constantin, L.A., Zhou, X., Burke, K.: Phys. Rev. Lett. 100, 136406 (2008)
Harb, M., Masih, D., Takanabe, K.: Phys. Chem. Chem. Phys. 16, 18198 (2014)
Acknowledgments
The authors are thankful for the support from the National Natural Science Foundation of China under Grant Nos. 51562021 and 11464027, the National Natural Science Foundation of Gansu Province under Grant Nos. 148RJZA027, the Program for Longyuan Youth Innovation Talents of Gansu Province of China, the Colleges and Universities Scientific Research Program of Gansu Province under Grant Nos. 2015B-040 and 2015B-048, and the Young Scholars Science Foundation of Lanzhou Jiaotong University under Grant No. 2014022.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Tian, JH., Sun, XW., Song, T. et al. Ferromagnetism and the Optical Properties of Mn-Doped CdSe with the Wurtzite Structure. J Supercond Nov Magn 30, 3109–3115 (2017). https://doi.org/10.1007/s10948-017-4109-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10948-017-4109-2