
Abstract
First-principles density functional theory (DFT) calculations with the generalized gradient approximation (GGA) have been performed to investigate electronic band structures and magnetic and half-metallic characteristic of Ti2CoAl1−x Sn x (x = 0, 0.25, 0.50, 0.75, 1). The optimized equilibrium lattice constants were found to be equal to 6.08 and 6.37 Å for Ti2CoAl and Ti2CoSn, respectively. These ternary Heusler compounds are found to be a complete half-metal with a total magnetic moment of 2 and 3 μ B, respectively, which is in good compliance with the Slater–Pauling rule, Mtotal = Z total − 18. The spin-polarized band structures and density of states (DOS) of the Ti2CoAl1−x Sn x (0.25, 0.50, and 0.75) Heusler alloys confirm that they are half-metal due to the presence of the energy gap in the minority spin. Consequently, Ti2CoAl1−x Sn x (x = 0, 0.25, 0.50, 0.75, 1) alloys are predicted to be a promising candidate for the practical applications in spintronic devices.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Heusler alloys with the general chemical formula X2YZ, where the X and Y atoms are transition metals and Z is a nonmagnetic metal or a nonmetallic element, have been attracting considerable interest due to their unique transport and magnetic properties [1], and they are the objects of great attention of researchers. In addition, they also serve as a source of new ideas and concepts on the formation of the electronic structure of magnets [2] and other spin-polarized compounds. Heusler alloys are known to be the essential ingredient for spintronic devices because some of them have been predicted to demonstrate half-metallicity, which maximizes the spin injection rate from ferromagnetic to semiconducting materials [3]. Half-metallic ferromagnets (HMFs) have shown great potential in spintronic devices since their introduction by de Groot and colleagues in the early 1980s [4]. Ideal half-metals (HMs) exhibit no energy bandgap for one spin channel, while indicating semiconductor behaviour for the other spin channels at the Fermi level. Due to the gap for one spin direction, the density of states at the Fermi level has 100 % theoretical spin polarization. The NiMnSn with the C1b structure is the first predicted HM ferromagnet [5]. In recent years, half-metallic ferromagnetism has been investigated for X2MnGe (X = Sc, Fe, Ni) [6], Ti2ZAl (Z = Co, Fe, Mn) [7], Co2CrX (X = Al, Ga, In) [8], Co2MnZ (Z = Al, Ge, Si, Ga) [9], and Fe2XAl (X = Cr, Mn, Ni) [10].
A few studies have been performed for the Ti2-based HM Heusler alloys. Feng et al. [11] used first-principles calculations to show that the full-Heusler compound Ti2NiAl with a Hg2CuTi-type structure is a new HM ferromagnet. Kervan et al. studied the electronic and the magnetic properties of the full-Heusler Ti2CoGa [12], Ti2CoAl [13], Ti2FeSi [14], and Ti2CoB [15] compounds. They predicted that all these compounds were HM ferrimagnets with a total magnetic moment of 2 μ B and that a complete spin polarization (100 %) exists around the Fermi level at the equilibrium lattice constant. Recently, Zheng and Jin [16] explored the half-metallicity of full-Heusler Ti2YAl (Y = V, Cr, Mn, Fe, Co, Ni, Cu, and Zn) alloys with the CuHg2Ti-type structure, and they showed that the Ti2MnAl alloy was a HM antiferromagnet, the Ti2YAl (Y = Fe, Co, and Ni) alloys were HM ferrimagnets, and the Ti2YAl (Y = V, Cr, Cu, and Zn) alloys were conventional ferromagnets [17]. In this paper, we present a study on the Ti2CoAl1−x Sn x (x = 0, 0.25, 0.50, 0.75, 1) Heusler alloys. In these alloys, up to now, no reports on the half-metallicity have been reported for x = 0.25, x = 0.50, and x = 0.75. Therefore, it is necessary to systematically study the structural, electronic, and magnetic properties and the half-metallic behaviour of the Ti2CoAl1−x Sn x (x = 0, 0.25, 0.50, 0.75, 1) alloys by the density functional calculations. This paper is structured as follows, where a brief description of the theoretical method and the different parameters used in our calculations is presented in Section 2. Through Section 3, we discuss the ground-state properties of the considered ternary compounds and their quaternary as well as their electronic band structures and magnetic properties. Finally, the results are summarized in Section 4.
2 Computational Details
The calculations of the present study are performed within a framework of the density functional theory (DFT) [18, 19]. We have utilized the full-potential linearized augmented plane wave (FP-LAPW) method as implemented in the WIEN2k code [20], which is one of the most accurate methods of simulating and calculating the ground-state properties of crystalline materials [21]. The exchange correlation term has been considered within the generalized gradient approximation (GGA) [22] to study the electronic structure and magnetic properties of Ti2CoAl1−x Sn x (x = 0, 0.25, 0.50, 0.75, 1). The basic functions are expanded into spherical harmonic functions and Fourier series inside the muffin-tin sphere and in the interstitial regions, respectively. The muffin-tin sphere radii were 2.37 a.u. for Ti and Co and 2.23 a.u. for Al in Ti2CoAl and were 2.46 a.u. for Ti and Co and 2.31 for Sn in Ti2CoSn, respectively. The maximum value of the angular momentum (\(l_{\max }~=\) 10) is taken for the wave function expansion inside the muffin-tin spheres. The convergence of the basis set was controlled by a cutoff parameter, \(R_{\text {MT}}\cdot K_{\max }~=\) 7, where R MT is the smallest muffin-tin sphere radius and \(K_{\max }\) is the largest reciprocal lattice vector used in the plane wave expansion within the interstitial region. The cutoff energy, which defines the separation of the valence and core states, was chosen as 6 Ry. We select the charge convergence as 0.0001e during the self-consistency cycles. The magnitude of the largest vector in the charge density Fourier expansion was \(G_{\max }~=\) 14 (a.u.) −1. A mesh of 64 special Brillouin zone (BZ) k-points was used in the irreducible wedge of the BZ. The total energy dependence on the cell volume is fitted to the Murnaghan equation of state [23] to determine the ground-state properties, as shown in (1)
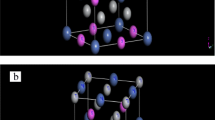
where E 0 is the minimum energy at T = 0 K, B is the bulk modulus, \(B\prime \) is the bulk modulus derivative, and V 0 is the equilibrium volume. In order to simulate the Ti2CoAl1−x Sn x (x = 0.25, 0.50, 0.75) quaternary alloy, we have used a supercell with 16 atoms. For x = 0.25, we substituted one atom of Al by Sn; for x = 050, we substituted two atoms of Al by two atoms of Sn; and for x = 0.75, we substituted three atoms of Al by three atoms of Sn. The crystal structure of Ti2CoAl1−x Sn x (x = 0, 0.25, 0.50, 0.75, 1) is shown in Fig. 1.

Crystal structure of the Ti2CoAl1−x Sn x crystalline alloys (x = 0, 0.25, 0.50, 0.75, 1)
3 Results and Discussion
In this section, we present the results of the geometrical structure optimization for the Ti2CoAl1−x Sn x (x = 0, 0.25, 0.50, 0.75, 1) Heusler alloys in addition to the lattice parameters and bulk modulus. The variation of the total energy with the volume is fitted to the Murnaghan equation of state [23] to obtain the equilibrium lattice constant and bulk modulus. The X2YZ Heusler compounds, which crystallize in the cubic L21 structure, have two types: the Cu2MnAl and Hg2CuTi structures. The two structures consist of four inter-penetrating Fcc sublattices, which have four unique crystallites. In the case of the Cu2AlMn-type L21 structure, the sequence of the atoms occupying the four sites of the unit cell is X–Y–X–Z, where the X atoms occupy positions A (0, 0, 0) and C (1/2, 1/2, 1/2) and the Y and Z atoms are located at B (1/4, 1/4, 1/4) and D (3/4, 3/4, 3/4), respectively. While for the Hg2CuTi-type L21 structure, the Y and the second X exchange sites and the sequence of the atoms is X–X–Y–Z, where the X atoms occupy the A (0, 0, 0) and B (1/4, 1/4, 1/4) sites, the Y atom occupies the C (1/2, 1/2, 1/2) site, and the Z atom occupies the D (3/4, 3/4, 3/4) site, respectively. The equilibrium lattice constants, bulk modulus, and these first derivatives are presented in Table 1. The optimized lattice constants for the Ti2CoAl and Ti2CoSn alloys are 6.08 and 6.37 Å, respectively, and they are in fairly good accordance with the previous theoretical calculations of 6.14 Å [13, 16] and 6.36 Å [17]. To the best knowledge of the author, there are no comparable studies about Ti2CoAl1−x Sn x (x = 0.25, 0.50, and 0.75) in the literature. Hence, we estimated the lattice parameters by Vegard’s law in Eq. (2)
For the Ti2CoAl and Ti2CoSn inverse Heusler alloys, the spin-polarized band structure and the total density of states (DOS) are depicted in Figs. 2 and 3, respectively. It is clear from Fig. 2 that the majority-spin oriented bands are metallic while the minority-spin oriented band shows a semiconducting gap around the Fermi level. This energy gap in the minority-spin band leads to almost 100 % spin polarization at the Fermi level, and this energy gap can be calculated using the energies of the highest occupied band at the Γ point and the lowest unoccupied band at the X point.

The spin-polarized band structure of Ti2CoAl and Ti2CoSn

DOS of Ti2CoAl and Ti2CoSn in the Hg2CuTi-type structures
Figure 3 shows a minority-spin channel where the total DOS in the region of the Fermi level is mainly due to the presence of the d electrons of the Ti(1), Ti(2), and Co transition metals. One can see the presence of the majority-spin states at the Fermi level and a wide bandgap in the minority-spin state confirming the HM characteristic of Ti2CoAl and Ti2CoSn. Within the −5 to −1 eV energy range, the s electrons of Al are contributed prevailingly. The minority-spin bandgap is an essential factor in the HM materials, and the cause of the HM bandgap is discussed as follows. The HM bandgaps usually take place from three aspects [17]: the covalent bandgap which exists in the half-Heusler alloy with the C1b structure, the d–d bandgap that is originated from the HM bandgap in the full-Heusler alloys with the AlCu2Mn structure, and finally, the charge transfer bandgap [17] which is usually observed in CrO2 and double perovskites [17]. For the Ti2CoAl and Ti2CoSn crystalline alloys with the Hg2CuTi-type structure, the Ti atoms occupy two sublattices with different coordination environments. In addition, the Ti2CoAl and Ti2CoSn alloys with the Hg2CuTi-type structure have a space group which is similar to that one of the half-Heusler alloys, but with an additional occupied site. Therefore, investigation of the origin of the bandgap in the Hg2CuTi-type structure of the Ti2CoAl and Ti2CoSn alloys is important [25].
The origin of the bandgap in Ti2FeSn, for example, has been discussed by Ahmadian [25]: Ti(1) and Ti(2) atoms have tetrahedral T d symmetry, and the d orbital of these atoms is divided into doubly and triply degenerated orbital, i.e., e g (d x2−y2, d z2) and t 2g (d xy, d yz, d zx) orbitals. A strong hybridization exists between the t 2g (e g) states of Ti(1) and Ti(2), leading to bonding and antibonding of the e g and the t 2g states. Similarly, the Fe atom has a T d symmetry, and the d orbitals split into doubly degenerated e g (d x2−y2, d z2) and triply degenerated t 2g (d xy, d yz, d zx) states. The e g and the t 2g states of the Ti(1)–Ti(2) coupling are further hybridized with the e g and the t 2g states of Fe, which results in the bonding (e g, t 2g) and antibonding (e g, t 2g) states. At the same time, the Fermi level is located in the gap between the bonding t 2g and the antibonding t 2g states. Within the group theory symmetry analysis, the antibonding states of the Ti(1)–Ti(2) coupling cannot be hybridized to the e g and the t 2g states of Fe. Therefore, the d–d hybridization between transition metals can participate in the formation of the minority bandgap [25].
The spin-polarized band structure and the total DOS for Ti2CoAl1−x Sn x (x = 0.25, 0.50, and 0.75) are presented in Figs. 4 and 5, and we can observe that E F cuts all the way through the bands in the majority-spin channel, which is the spin-up case, while the energy gap of Ti2CoAl1−x Sn x (x = 0.25, 0.50, and 0.75) resides in the minority-spin channel (spin-down). This means that the majority-spin states have a metallic character, while the minority-spin band contains an energy gap at the Fermi level (E F), which is a semiconducting one. This suggests that Ti2CoAl1−x Sn x (x = 0.25, 0.50, and 0.75) are half-metals and electrons at the Fermi level are 100 % spin-polarized. It is necessary to emphasize the substantial anisotropy of the electron bands for two directions: R–X–M and G–X. This factor may be indicated on different hole mobilities in these directions, particularly in the R–X–M BZ direction as there are more delocalized band carriers and in the G–X direction as there are more localized states.

The spin-polarized band structure of Ti2CoAl1−x Sn x (x = 0.25, 0.50, 0.75)

DOS of Ti2CoAl1−x Sn x (x = 0.25, 0.50, 0.75)
Slater and Pauling discovered that the magnetic moment (m) for the 3d elements and their binary alloys can be estimated on the basis of the average valence electron number (N V) per atom [26, 27]. The materials are divided into two areas depending on the magnetic moment near N V: The first area of the Slater–Pauling curve is the area of low-valence electron concentrations (N V ≤ 8) and of localized magnetism. The second area is the area of high-valence electron concentrations (N V ≥ 8) and of itinerant magnetism where the magnetic moment is in multiples of Bohr magnetons (μ B) given by m = N V−2n ↓, where 2n ↓ denotes the number of electrons in the minority-spin states. The half-metallic materials obey the Slater–Pauling rule [26, 27]: m HMF≈N V−6, where m HMF is the mean magnetic moment per atom. In the case of the compounds having four atoms per unit cell, one has to subtract 24 (6 multiplied by the number of atoms) from the accumulated number of valence electrons in the unit cell N V to find the spin magnetic moment per unit cell: m HMF = N V−24.
Recent research reveals that many Ti2-based full-Heusler alloys with the Hg2CuTi-type structure can also belong to the family of half-metallic material, and the total magnetic moments of these alloys follow the Mtotal = Ztotal − 18 rule instead of the Mtotal = Ztotal − 24 rule [13]. The Ti2CoAl alloy has 20 valence electrons per unit cell (Ztotal = 20), and therefore, the spin magnetic moment is 20 − 18 = 2 μ B for each unit cell. The Ti2CoSn alloy has 21 valence electrons per unit cell, so the spin magnetic moment is 21 − 18 = 3 μ B.
The calculated total magnetic moment of the Ti2CoAl compound is 2 μ B (as shown in Table 2), while the atomic magnetic moments are 1.14 μ B for Ti(1), 0.65137 μ B for Ti(2), −0.29248 μ B for Co, 0.00109 μ B for Al, and 0.49423 μ B for Sn, respectively, for the interstitial moment. For Ti2CoSn, the total magnetic moment is 3 μ B with the atomic magnetic moments: 1.41715, 0.73736, 0.36232, −0.00594, and 0.48920 μ B for Ti(1), Ti(2), Co, Sn, and interstitial moments, respectively. The atom magnetic moments for Ti2CoAl in the present study are in good agreement with the results reported by Zheng et al. [16] and Bayar et al. [13]. The calculated integer magnetic moment of the Ti2CoAl and Ti2CoSn compounds indicates the stability of half-metallic behaviour. It is seen that the total magnetic moment per unit cell is increased as a function of x concentration (2 μ B, 9 μ B/16 atoms, 10 μ B/16 atoms, 11 μ B/16 atoms, and 3 μ B, for x = 0, 0.25, 0.50, 0.75, and 1, respectively), for Ti2CoAl1−x Sn x (x = 0, 0.25, 0.50, 0.75, 1). It can be seen from Table 2 that the different local magnetic moments for the two Ti atoms in the Ti2CoAl and Ti2CoSn alloys with the CuHg2Ti-type structure originated from different atomic environments, i.e., the Ti(1) atom has four nearest Ti(2) atoms and four nearest Al (Sn) atoms as well as six second-nearest Co atoms, while the Ti(2) atom has four nearest Ti(1) atoms and four nearest Co atoms as well as six second-nearest Al (Sn) atoms.
The presented results may be helpful for the analysis of magnetic spin polarization for other half-metallicity in Heusler alloys such as Zr2VZ (Z = Al, Ga, In) [28], Ti2YPb (Y = Co, Fe) [29], and Mn2ZrSi and Mn2ZrGe [30]. All these compounds are characterized by an enhanced number of defect states which may change experimentally observed magnitudes of magnetization.
4 Conclusions
For the Ti2CoAl1−x Sn x (x = 0, 0.25, 0.50, 0.75, 1) Heusler alloys, the electronic structure and magnetic properties have been calculated using the first-principles FP-LAPW method. The spin-polarized calculations showed that the Ti2CoAl and Ti2CoSn Heusler compounds are a half-metallic with a magnetic moment of 2 and 3 μ B, respectively. Ti2CoAl1−x Sn x (x = 0, 0.25, 0.50, 0.75, 1) indicates that the quaternary Heusler alloys have a perfect half-metallic character with semiconducting minority-spin band structure and 100 % polarization. Consequently, Ti2CoAl1−x Sn x (x = 0, 0.25, 0.50, 0.75, 1) is predicted to be a good promising candidate to explore the half-metallic ferromagnetism for the practical applications of spintronic devices. The calculations have shown a substantial anisotropy of electron bands for two BZ directions: R–X–M and G–X. This factor may be indicated on different hole mobilities in these directions, particularly in the R–X–M direction where there are more delocalized band carriers and in the G–X direction where there are more localized states. This may be used for the creation of different spintronic devices.
References
Fukatani, N., Fujita, H., Miyawaki, T., Ueda, K., Asano, H.: J. Kor. Phys. Soc. 63(3), 711–715 (2013)
Shreder, E.I., Svyazhin, A.D., Fomina, K.A.: Phys. Met. Metallogr. 113, 146 (2012)
Quang, T.V., Lee, J.I., Kim, M.: J. Kor. Phys. Soc. 62(12), 2184–2187 (2013)
de Groot, R.A., Mueller, F.M., van Engen, G.P., Buschow, K.H.J.: Phys. Rev. Lett. 50, 2024 (1983)
Wei, X.-P., Chu, Y.-D., Sun, X.-W., Deng, J.-B.: Eur. Phys. J. B 86, 450 (2013)
Dahmane, F., Mesri, D., Doumi, B., Tadjer, A., Abbar, B., Yakoubi, A., Boutaleb, M., Aourag, H.: J. Supercond. Nov. Magn. doi:10.1007/s10948-015-2994-9
Dahmane, F., Benalia, S., Djoudi, L., Tadjer, A., Khenata, R., Doumi, B., Aourag, H.: J. Supercond. Nov. Magn. doi:10.1007/s10948-015-3109-3
Dahmane, F., Mesri, D., Tadjer, A., Khenata, R., Benalia, S., Djoud, L., Doumi, B., Boumia, L., Aourag, H.: Mod. Phys. Lett. B 30, 1550265 (2016)
Dahmane, F., Doumi, B., Mogulkoc, Y., Tadjer, A., Prakash, D., Verma, K.D., Varshney, D., Gheboul, M.A., Bin Omran, S., Khenata, R.: J. Supercond. Nov. Magn. doi: doi:10.1007/s10948-015-3357-2
Dahmane, F., Mogulkoc, Y., Doumi, B., Tadjer, A., Khenata, R., Bin Omran, S., Rai, D.P., Murtaza, G., Varshney, D.: J. Magn. Magn. Mater. 407, 167 (2016)
Feng, L., Tang, C., Wang, S., He, W.: J. Alloy. Comp. 509, 5187 (2011)
Kervan, N., Kervan, S.: J. Magn. Magn. Mater. 324, 645 (2012)
Bayar, E., Kervan, N., Kervan, S.-U.: J. Magn. Magn. Mater. 323, 2945 (2011)
Kervan, N., Kervan, S.: J. Phys. Chem. Solids 72, 1358 (2011)
Kervan, N., Kervan, S.: Solid State. Commun. 151, 1162 (2011)
Zheng, N., Jin, Y.: J. Magn. Magn. Mater. 324, 3099 (2012)
Ahmadian, F.: J. Alloys Compd. 576, 279 (2013)
Hohenberg, P., Kohn, W.: Phys. Rev. B 136, 864 (1964)
Kohn, W., Sham, L.J.: Phys. Rev. A 140, 1133 (1965)
Blaha, P., Schwarz, K., Madsen, G.K.H., Kvasnicka, D., Luitz, J.: WIEN2k, an Augmented Plane Wave Plus Local Orbitals Program for Calculating Crystal Properties. Vienna University of Technology, Vienna (2001)
Wong, K.M., Alay-e-Abbas, S.M., Fang, Y., Shaukat, A., Lei, Y.: J. Appl. Phys. 114, 034901 (2013)
Perdew, J.P., Burke, K., Ernzerhof, M.: Phys. Rev. Lett. 77, 3865 (1996)
Murnaghan, F.D.: Proc. Natl. Acad. Sci. USA 30, 5390 (1944)
Wei, X.P., Deng, J.B., Mao, G.Y., Chu, S.B., Hu, X.-R.: Intermetallics 29, 86 (2012)
Ahmadian, F.: J. Kor. Phys. Soci. 64, 277 (2014)
Slater, J.C.: Phys. Rev. 49, 537 (1936)
Pauling, L.: Phys. Rev. 54, 899 (1938)
Gao, Y.C., Wang, X.T., Rozale, H., Lu, J.W.: J. Kor. Phys. Soc. 67, 881 (2015)
Hussain, M.K., Gao, G.Y., Yao, K.-L.: Int. J. Mod. Phys. B 29, 1550175 (2015)
Abada, A., Amara, K., Hiadsi, S., Amrani, B.: J. Magn. Magn. Mater 388, 59 (2015)
Acknowledgments
The authors Khenata and Bin Omran extend their sincere appreciation to the Deanship of Scientific Research at King Saud University for funding this Prolific Research Group (PRG-1437-39).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Dahmane, F., Khenata, R., Doumi, B. et al. Band Structure Simulations of the Structural, Electronic, Magnetic, and Half-Metallic Features of the Ti 2 CoAl1−x Sn x (x = 0, 0.25, 0.50, 0.75, 1) Heusler Alloys. J Supercond Nov Magn 29, 3193–3199 (2016). https://doi.org/10.1007/s10948-016-3711-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10948-016-3711-z