
Abstract
Nanocrystalline Fe90Ni10 alloys were synthesized by mechanical alloying, starting from a powder mixture of elemental Fe and Ni. The phase evolution and magnetic properties were investigated, as a function of milling time, using the X-ray diffraction (XRD), the vibrating sample magnetometer (VSM), and the 57Fe Mössbauer spectroscopy. From XRD results, we concluded the formation, after 13 h of milling, of a disordered phase α-Fe(Ni) (bcc). It has been shown that the increase of milling time decreases the crystallites size and increases the microstrains and the lattice parameter. When the crystallites size decreases, the coercive field, H c, decreases first, then increases and finally reaches a constant value of about 26 Oe. During the periode of the alloy formation, the saturation magnetization, M s, increases with decreasing crystallite size and reaches the highest value of 212 emu/g after 27 h of milling, then, M s remains constant up to 48 h of milling. The adjustment of Mössbauer spectra revealed that the fraction of the (bcc) α-Fe(Ni) phase increased with milling time. After 13 h of milling, only the (bcc) α-Fe(Ni) phase is observed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Iron-based alloys have received appreciable attention due to their remarkable soft magnetic properties, such as high saturation magnetization and high permeability [1]. Most recently, research interest in nanocrystalline materials has dramatically increased because of their attractive properties especially magnetic ones often superior to those of conventional polycrystalline materials [2]. Those properties arise from the small crystallite size and a significantly large volume of crystallite boundary [3]. Among different iron-based nanocrystalline soft ferromagnetic alloys, the Fe–Ni system has been studied for a long time due to its excellent soft magnetic performances that make them of potential use in various applications such as in catalysis, sensors, disk drive components, and absorbing materials [4–10]. A variety of processing techniques has been developed for the production of nanocrystalline Fe–Ni including chemical processing, sol–gel methods, rapid solidification, electrodeposition, and ball milling [11–19]. Mechanical alloying (MA) is a solid-state powder processing technique that can be used to synthesize a wide variety of nanocrystalline phases from elementary powders [20, 21]. During MA, the powder particles are continuously subjected to severe plastic deformations and fractures, leading to the incorporation of lattice defects and to a continuous refinement of the initial structure [22]. It was generally demonstrated that the magnetic properties of mechanically alloyed materials can be notably improved when the crystallite size is reduced into the nanometer scale [23, 24]. In the last years, many studies have focused on the Fe–Ni powders obtained by MA. It has been shown that different structures were formed, depending on chemical composition of this alloy. In this regard, S.D. Kaloshkin et al. [25] investigated by MA the phase formation in Fe 100− x Ni x alloys (10 < x < 90 at.%). They reported that for the concentrations lower than 20 at.%, only single cubic-centered (bcc) phase is present. Increasing the concentration of Ni up to 28 at.% leads to the formation of both cubic-centered (bcc) and face-centered cubic (fcc) phases. Only (fcc) phase has been evidenced in the samples when the concentrations of Ni is higher to 28 at.%. J. Liu et al. [26] concluded the formation of the single (fcc) NiFe phase in Fe–40 wt.%Ni alloy after 160 h of milling. A. Guittoum et al. [27] and Kh. Gheisari et al. [28] obtained nanocrystalline (fcc) phase by MA of Fe–50 at.%Ni powders. On the other hand, the magnetic properties of mechanically alloyed Fe–Ni alloys presented a great interest for researchers. The basic premise has been that the magnetic behavior is intimately related to the structure but not notably affected by the milling conditions. For example, R. Hamzaoui et al. [17] showed that the saturation magnetization, M s, of the nanocrystalline Fe 90Ni 10 and Fe 80Ni 20 (wt.%) alloys prepared by high-energy ball milling is not modified by the milling mode process; nevertheless, M s is appreciably improved by the decrease of the crystallite size and the increase of the lattice parameter. More recently, R.R. Rodriguez et al. [19] have investigated, by means of X-ray diffraction (XRD) and Mössbauer spectroscopy (MS) at low temperature, the structural and hyperfine properties of nanostructured Fe 65Ni 35 alloys. The same authors [19] reported that the XRD results showed the coexistence of one (bcc) and two (fcc) phases. According to MS results, the (bcc) phase was identified as a ferromagnetic-disordered Fe(Ni) phase rich in Fe, while the (fcc) phases were attributed, respectively, to the ferromagnetic-disordered phase rich in Ni and to the paramagnetic one rich in Fe.
In a perspective of developing more detailed study on Fe–Ni system, we have performed this research work which treats the structural and magnetic properties of Fe 90Ni 10nanostructured powders elaborated by high-energy ball milling for different milling times (up to 48 h). In order to well characterize the elaborated Fe 90Ni 10samples, many methods have been used in this work: the X-ray diffraction (XRD) followed by the Rietveld refinement to study the microstructural change occurring after different milling times, the vibrating sample magnetometry (VSM) which allows us to study the behavior of the saturation magnetization and the coercive field with the crystallite size, and finally the Mössbauer spectroscopy (MS) which enables us to follow the structural changes at an hyperfine level.
2 Experimental Procedures
2.1 Elaboration of Fe90Ni10 Nanostructured Powders
The starting elemental powders of Fe and Ni (purity >99.9 %, average particle size <100 μm) were weighed and mixed to give the composition of Fe 90Ni 10 (at.%). The mechanical alloying experiments were performed using a vario-planetary high-energy ball mill (Fritsch P4). The advantage of the Fritsch P4 remains in the possibility of choosing the milling mode, i.e., the rotational speeds of the disk (ω) and the vials (Ω) are independent. In order to ensure that the milling will be performed in an inert atmosphere (no oxidation during the milling), the starting powders with five (05) stainless steel balls, having 12 mm in diameter, were sealed in atmosphere of argon in a stainless steel vial. The ball to powder weight ratio was fixed to 15:1, and the milling process was carried out with a sequence of 30 min of milling following by 15 min of pause. The rotation speeds of disc and vials were 250 and 500 rpm, respectively, and were maintained for the different milling times ranging from 4 to 48 h.
2.2 Samples Characterization
The X-ray diffraction experiments were performed with a Philips X’Pert Pro diffractometer working with the Bragg–Brentano geometry using CuK α radiation. The XRD patterns were collected in the 2 𝜃 angular range from 20∘ to 110∘ with a step size of 0.02 ∘ . The MAUD software (version 2.33), based on the Rietveld method combined with Fourier analysis [29–31], was used to refine the microstructural parameters of nanostructured powders. The instrumental resolution of the X-ray diffraction equipment was determined with a standard sample of silicon (Si) free from defects. The background of each X-ray pattern was fitted by a polynomial function, and the microstructural parameters such as crystallite size and microstrain were obtained after the adjustment of the spectra by applying the Delf isotropic model [32]. To do so, the observed lines profiles are adjusted with a pseudo-Voigt function, based on Caglioti formulas (see Eqs. (1) and (2)), and which is used by the most popular Rietveld refinement programs [32–35]. In this function, the full width at half maximum (FWHM) of the Gaussian (H G) and Lorentzian (H L) components of the line profile have an angular dependence given by
Here, U,V,W,X, and Y are refinable parameters, which depend on FWHM. Because the parameters in Eqs. (1) and (2) are depending of FWHM, they should be converted into integral breadth profile according to conversion factors given by Langford [36]. After removing the instrumental contribution, the integral breadths β D (microstrains effect) and β S (size effect) can be related to mean values of the crystallite size, < D>, and the root mean square (r.m.s) of the microstrains, \(\left \langle \sigma ^{2}\right \rangle ^{\frac {1}{2}}\), according the following equations [36–40]:
Here, λ is the X-ray wavelength in nanometer (nm), 𝜃 is Bragg’s diffraction angle, and K is a constant which depends on the crystallite shape.
The Mössbauer spectra were collected at room temperature (RT) using a Wissel instrument in the constant acceleration mode, using a radioactive 57Co source diffused into a rhodium matrix. Metallic iron was used for energy calibration and also as a reference for isomer shift. The adjustment of Mössbauer spectra was done with the Recoil software using the Voigt-based hyperfine field distribution method (HFD-VB-F) [41]. The hysteresis loops of the powder samples were measured at room temperature with a LakeShore 7400 vibrating scanning magnetometer (VSM) in a maximum applied magnetic field, H, of 20 kOe.
3 Results and Discussions
3.1 Microstructural Study
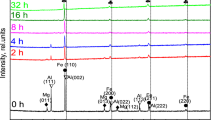
Figure 1 shows the XRD spectra obtained for different milling times. The XRD spectrum of the unmilled powder is also presented as a reference. For the unmilled sample, the XRD spectrum reveals the presence of both (bcc) α-Fe and (fcc) Ni in their elemental forms. The Ni (111) and Ni (222) peaks overlap with Fe (110) and Fe (220), respectively. After 4 h of milling, the intensities of Ni peaks intensely decreased indicating that the solid-state reaction starts in this step of milling process. The Rietveld refinement has been performed for all the XRD spectra of the milled powders. In Fig. 2, we have presented an example of refinement for the powder milled at 13 h. We have shown that, after 13 h of milling, no reflections corresponding to (fcc) Ni and (bcc) α-Fe exist, and that only the (bcc) α-Fe(Ni) phase with a space group Im3m is present. This result agrees well with the previous one reported by E. Jartych et al. [42], where a (bcc) α-Fe(Ni) solid solution was evidenced after 20 h of milling. Moreover, in addition to the complete disappearance of Ni peaks, a slight shift towards the lower angles which is accompanied with a broadening of α-Fe peaks have been observed (see Fig. 1). This behavior has been shown in different Fe–Nibased alloys and it has been indicated that the slightly angular shift is due to the relaxation of first-order internal stress during milling [27]. In fact, the first-order internal stresses acts at the macroscopic level by modifying the lattice parameter and consequently produce an angular shift of the XRD peaks [43]. On the other hand, the broadening of the peaks is attributed to the reduction in crystallite size and the increase in internal microstrains [44].

X-ray diffraction patterns of Fe 90Ni 10 milled powders for different times

Rietveld refinement of XRD patterns corresponding to the sample milled for 13 h
Figure 3 illustrates the variation of the lattice parameter, a(nm), of Fe 90Ni 10powders as a function of milling time. We remark that a(nm) increases slightly from its initial value of 0.2866 ± 0.0002 (corresponding to that of pure (bcc) α-Fe) to 0.2871 ± 0.0002 nm after 13 h of milling which corresponds to 0.17 % of lattice parameter expansion. With prolonging the milling time above 13 h, where only the (bcc) α-Fe(Ni) phase exists, the lattice parameter remains constant. The value of a(nm) obtained in this work agrees well with that reported in the literature for the ball milled Fe 90Ni 10 (wt.%) alloy, where the lattice parameter value obtained after 60 h of milling was 0.28695 ± 0.00005 nm [17]. During MA, the increase/decrease of a(nm) is generally attributed to the size mismatch between the solvent and solute atoms. However, for the Fe–Ni alloys, the difference between the atomic radii of Ni (R Ni = 0.125 nm) and Fe (R Fe = 0.124 nm) is very small. Then, we believe that, in our case, the increase of lattice parameter is not due solely to the diffusion of the Ni atoms into the α-Fe lattice. The lattice parameter expansion may be due also to the high density of defects induced by the ball milling process in the interfaces [45, 46].

Evolution of lattice parameter, a, as a function of milling time
Figure 4 presents the variation of the average crystallite size, < D>(nm), and the root mean square (r.m.s.) of the microstrains, \(\left \langle \sigma ^{2} \right \rangle ^{\frac {1}{2}}\), which have been deduced from the Rietveld refinement of XRD spectra. The estimation of errors in < D>and \(\left \langle \sigma ^{2} \right \rangle ^{\frac {1}{2}}\) is governed by the precisions in the determination of integral breadths (βs and β D) and the angular position of the diffraction peaks [47, 48]. These errors in < D>and \(\left \langle \sigma ^{2} \right \rangle ^{\frac {1}{2}}\) are given by the program Maud after performing a good fit (quality factor of fit, GoF, close to 1).

Evolution of the average crystallite size, < D>(nm), and root mean square (r.m.s.) of the microstrains, \(\left \langle \sigma ^{2} \right \rangle ^{\frac {1}{2}}\), as a function of milling time for Fe 90Ni 10 powders
From Fig. 4, one can see that < D>decreases sharply in the early stages of milling (0–13 h) and tends to saturate after 13 h at about 31 ± 2 nm. It is interesting to note that the value of < D>achieved in this work is highest to that obtained by R. Hamzaoui et al. [49] and lowest to the one found by R. Koohkan et al. [50]. The reduction of < D>is accompanied by a monotonic increase of \(\left \langle \sigma ^{2} \right \rangle ^{\frac {1}{2}}\) from its initial value of 0.05 ± 0.03 to 0.47 ± 0.03 % after 18 h of milling. With further milling time, it can be seen that \(\left \langle \sigma ^{2} \right \rangle ^{\frac {1}{2}}\) tends to be balanced around this value. This result agrees with that found by R. Hamzaoui et al. [49] for the Fe 90Ni 10 (wt.%) compound prepared by mechanical alloying. The decrease of < D>and the increase of \(\left \langle \sigma ^{2} \right \rangle ^{\frac {1}{2}}\) can be explained by the fact that, during high-energy ball milling, the powder particle are continuously subjected to severe plastic deformation and fracturing that results in the refinement of crystallite size as well as increase of the lattice microstrains [22].
3.2 VSM Measurement
Figure 5 shows the room temperature hysteresis loops (M−−H) of Fe 90Ni10 powders for the different milling times. From these curves, the values of saturation magnetization, M s, and coercivity, H c, have been computed. All the samples are ferromagnetic with a value of the coercivity, H c, below 30 Oe for all the samples then, we can consider these alloys as soft magnetic materials.

a Hysteresis loops (M– H curves) at room temperature of the Fe 90Ni 10 powders milled for a 0, 4, 13, 18, 27, 36, and 48 h; b zoom of the low applied magnetic field region
The evolution of H c as a function of crystallite size is shown in Fig. 6. As can be seen, H c does not follow a clear trend with the crystallite size. Indeed, we remark that at the first stage corresponding for milling time up to 13 h, H c decreases with decreasing crystallite size from 49 Oe (D = 154 nm) to 13.5 Oe (for D = 32 nm). Similar behavior was observed for nanocrystalline alloys elaborated by MA [51]. This decrease of coercivity when the crystallite size decreases can be due to the predominance of size effect compared to that of microstrains [51]. For the second stage (the milling time between 13 and 18 h) and for practically the same crystallite size value of around 31 nm, we remark that H c increases brutally and reaches a value of 26 Oe. The increase of H c may be due to the microstrain effect which seems to dominate in this range of the crystallite size [2].

Evolution of the coercive field, H c, as a function of the crystallite size
With prolonging milling time up to 48 h, we observed that H c remains constant for the same values of < D>(see Fig. 6). We believe that this behavior may be due to the fact that the balance between the two effects, i.e., the crystallite size decreases which tends to decrease coercivity and the effect of microstrains which tends to increase coercivity, has been achieved. Indeed, it was reported (see Section 3.1) that in this stage of milling, the mechanical alloying equilibrium and the complete formation of the alloy have been attained.
It is important to note, for comparison, that for Fe 90Ni 10 compound, R. Koohkan et al. [50] observed a behavior of H c different from that found in this work. The same authors [50] reported that the coercivity increases monotonically with decreasing crystallite size as a consequence of considerable introduction of internal strains into the material. Moreover, the same authors have obtained values of H c higher than those mentioned in the present study. We believe that such difference could be attributed to the conditions of preparation of samples.
Figure 7 shows the variation of the saturation magnetization as a function of crystallite size. It is observed a monotonic increase of M s, from 193 to 206 emu/g, when the crystallite size decreases from 154 to 31 nm. For 13 h of milling and for a constant value of crystallite size, we noted a sharp increase in M s which lasts up to 36 h. The maximum value achieved by M s is equal to 212 emu/g. A similar behavior was observed by many authors in the case of Fe–Ni alloys prepared by mechanical alloying [4, 49]. Generally, the increase of M s is attributed to the formation of (bcc) α-Fe (Ni) solid solution and also to the charge transfer between Fe and Ni atoms [1]. Moreover, the increase of M s can be also due to the diminishing of magnetocrystalline anisotropy as a result of the crystallite size decreasing, giving rise to an easier motion of the magnetization [7]. The value of M s obtained in this work is very close to that reported by J. Ding et al. [4] for the same composition but after 36 h of milling time.

Evolution of saturation magnetization, M s, as a function of crystallite size
3.3 Mössbauer Spectroscopy
Mössbauer spectroscopy is one of the most suitable methods which can be used to follow the phase evolution during the MA process. The room-temperature Mössbauer spectra of Fe 90Ni 10 milled powders and their corresponding hyperfine field distributions (HFDs) are shown in Fig. 8. All the hyperfine parameters resulting from the adjustment of the Mössbauer spectra are presented in Table 1. It is noted that, for 4 h of milling time, the HFD is characterized by a narrow peak with a small shoulder in the region of higher hyperfine magnetic fields (higher than 33 T corresponding to pure Fe). For this sample, the Mössbauer spectrum was fitted with one sextet containing two components. The first component having a mean hyperfine field value, < H hf>, of 32.97 T is a characteristic of the starting Fe powder where the Fe atoms are surrounded, preferentially, by Fe atoms as first nearest neighbors. The second component with < H hf>equal to 34.34 T is attributed to the formation of a disordered (bcc) α-Fe (Ni) phase as a result of the substitution of Fe atoms by Ni ones. This result correlates well with that found by XRD diffraction where the formation of the disordered (bcc) α-Fe(Ni) phase was clearly proved by the drastic diminution of the diffraction peaks intensity of (fcc) Ni phase after 4 h of MA (see Section 3.1). With increasing milling time until 9 h, one can see that the HFD presents a clear asymmetry. Indeed, the shoulder corresponding to the second component (bcc α-Fe (Ni)) becomes more intense and the intensity of the narrow pick corresponding to the first component (bcc α-Fe) has considerably decreased (see Fig. 8). For this sample, the value of < H hf>obtained for the first and the second component are 32.96 and 33.64 T, respectively. After 13 h of milling, we note that the HFD is highly symmetric, indicating that the sample presents a more homogeneous composition. For this sample, the spectrum was also adjusted with two components and the obtained < H hf>values were 33.09 and 33.75 T, respectively. No change was noted in the shape of the HFD when the milling time increased until 48 h. Indeed, the Mössbauer spectra consist only of one component characteristic of the disordered (bcc) α-Fe(Ni) phase where < H hf>is equal to 33.81, 33.79 T, 33.77, and 33.78 T for 18, 27, 36, and 48 h of milling, respectively.

Room-temperature Mössbauer spectra and their hyperfine magnetic field distribution of Fe 90Ni 10 powders milled for different durations
The evolution of the relative areas of the two components as a function of milling time is shown in Fig. 9. As the milling time increases, the relative area of the (bcc) α-Fe (Ni) phase gradually increases in relation with the progressive formation of the solid solution. The increase of the (bcc) α-Fe (Ni) relative area is accompanied with a decrease of that of (bcc) α-Fe. It is clearly observed that after 13 h of milling, the relative intensity of this contribution has considerably reduced (less than 5 %). One can conclude that the (bcc) α-Fe (Ni) solid solution is formed by the diffusion of Ni atoms into the Fe lattice.

Mössbauer relative areas of the (bcc) α-Fe and (bcc) α-Fe (Ni) as a function milling time
4 Conclusion
In the present work, we have prepared nanostructured Fe 90Ni 10 (at.%) powders using high-energy ball milling for different milling times up to 48 h. The samples were investigated by X-ray diffraction XRD, Mössbauer spectroscopy, and magnetic measurements. The XRD patterns reveal that the (bcc) α-Fe(Ni) solid solution is formed by the gradual diffusion of Ni atoms into (bcc) α-Fe. After 13 h of milling, only the (bcc) α-Fe(Ni) phase is detected. With increasing milling time, we noted that the lattice parameter increases slightly from its initial value corresponding to that of (bcc) α-Fe to its final value of 0.2871 nm. In addition, we recorded that the alloy formation was accompanied with a decrease of the crystallite size accompanied by an increase in microstrains, and the final values achieved are 31 nm and 0.47 %, respectively. As the crystallite size decreases, the saturation magnetization increased up to 27 h to reach a maximum value of 212 emu/g and then remained constant. The coercive field, H c, decreases first when the crystallite size decreases, then increases and reaches finally a constant value of 26 Oe. From Mössbauer results, we concluded that the alloy starts to form during the first hours of milling; however, after 13 h of milling, only the component corresponding to (bcc) α-Fe(Ni) phase is present.
References
Duan, Y., Zhang, Y., Wang, T., Gu, S., Li, X., Lv, X.: Evolution study of microstructure and electromagnetic behaviors of Fe-Co-Ni alloy with mechanical alloying, Matr. Sci. Eng: B 185, 86–93 (2014)
Sekri, A., Khitouni, M., Escoda, L., Suño, l.J.J., Dammak, M., Greneche, J.M.: Microstructure evolution and thermal stability of nanostructured Fe50Al30(Ni70Zr30)10B10 powders produced by mechanical alloying. Superlattices Microstruct 74, 156–166 (2014)
Koch, C.C.: Nanostructured Materials Processing, properties, and applications, Andrews Appl. Sci. Publ. New York (2007)
Ding, J., Shi, Y., Chen, L.F., Deng, C.R., Fuh, S.H., Li, Y.: A Structural, magnetic and microwave study on mechanically alloyed Fe-based alloy powders. J. Magn. Magn. Mater 247, 249–256 (2002)
Guittoum, A., Layadi, A., Taffat, H., Bourzami, A., Souami, N., Lenoble, O.: Structure, Mossbauer and magnetic studies of nanostructured Fe 80Ni 20 alloy elaborated by mechanical alloying. Philos. Mag. 88, 1085–1098 (2008)
Lee, G.Y., Kwon, S.K., Lee, J.S.: Annealing effect on microstructure and magnetic properties of flake-shaped agglomerates of Ni–20wt%Fe nanopowder. J. Alloys Compd 613, 164–169 (2014)
Guittoum, A., lamrani, S., Bourzami, A., Hemmous, M., Souami, N., Weber, W.: Elaboration, structure, microstructure and magnetic properties of soft Fe80Ni20 nanomaterials elaborated by high energy ball milling. Eur. Phys. J. Appl. Phys 65, 30401–30409 (2014)
Sun, Y., Liang, Q., Zhang, Y., Tian, Y., Liu, Y., Li, F., Fang, D.: Controlled synthesis of magnetic Ni–Fe alloy with various morphologies by hydrothermal approach. J. Magn. Magn. Mater 332, 85–88 (2013)
Kaibi, A., Guittoum, A., Fenineche, N., Souami, N., Kechouane, M.: Structural, microstructural and magnetic properties of nanocrystalline Ni 75Fe 25 compound produced by mechanical alloying. Sens. Lett 11, 1–7 (2013)
Feng, Y., Qiu, T.: Preparation, characterization and microwave absorbing properties of FeNi alloy prepared by gas atomization method. J. Alloys Compd 513, 455–459 (2012)
Moustafa, S.F., Daoush, W.M.: Synthesis of nano-sized Fe–Ni powder by chemical process for magnetic applications. J. Mater. Proc. Techn 181, 59–63 (2007)
Zhang, J., Jin, M., Xiao, F., Li, C., Jin, X.: Martensitic transformation in Fe–Ni nanoparticles prepared by a sol–gel process. Mater. Lett 80, 152–154 (2012)
Suñol, J.J., Gonzalez, A., Escoda, L.: Comparison of Fe-Ni based alloys prepared by ball milling and rapid solidification. J. mater. Sci 39, 5147–5150 (2004)
Zhang, B., Fenineche, N.E., Zhu, L., Liao, H., Coddet, C.: Studies of magnetic properties of permalloy (Fe–30%Ni) prepared by SLM technology. J. Magn. Magn. Mater 324, 495–500 (2012)
Ma, J., Qin, M., Zhang, L., Zhang, R., Tian, L., Zhang, X., Li, X.: Magnetic properties of Fe–50%Ni alloy fabricated by metal injection molding. Mater. Des. 51, 1018–1022 (2013)
Vitta, S., Khuntia, A., Ravikumar, G., Bahadur, D.: Electrical and magnetic properties of nanocrystalline Fe100−xNix alloys. J. Magn. Magn. Mater 320, 182-189 (2008)
Hamzaoui, R., Elkhedim, O., Gaffet, E.: Milling conditions effect on structure and magnetic properties of mechanically alloyed Fe–10% Ni and Fe–20% Ni alloys. Mater. Sci. Eng 381, 363–371 (2004)
Suñol, J.J., Saurina, J., Bruna, P.: Structural and thermal changes induced by mechanical alloying in Fe-Ni based amorphous melt-spun alloy. j. mater. Che 114, 996–999 (2009)
Rodriguez, R.R., Valenzuela, J.L., Tabares, J.A., Pérez Alcázar, G.A.: Mössbauer and X-ray study of the Fe 65Ni 35 invar alloy obtained by mechanical alloying. Hyperfine Interact 224, 323–330 (2014)
Krifa, M., Mhadhbi, M., Escoda, L., Saurina, J., Suñol, J.J., Llorca-Isern, N., Artieda-Guzman, C., Khitouni, M.: Phase transformations during mechanical alloying of Fe–30% Al–20% Cu. Powder Technol 246, 117–124 (2013)
Birringer, R.: Nanocrystalline materials. Mat. Sci. Eng. A 117, 33–43 (1989)
Suryanarayana, C.: Mechanical alloying and milling. Prog. Mater. Sci 46, 1–184 (2001)
Zeng, Q., Baker, I., Creary, V.M., Yan, Z.: Soft ferromagnetism in nanostructured mechanical alloying FeCo-based powders. J. Magn. Magn. Mater 318, 28–38 (2007)
Neamtu, B.V., Geoffroy, O., Chicinas, I., Isnard, O.: AC magnetic properties of the soft magnetic composites based on Supermalloy nanocrystalline powder prepared by mechanical alloying. Mat. Sci. Eng. B 177, 661–665 (2012)
Kaloshkin, S.D., Tcherdyntsev, V.V., Tomilin, I.A., Baldokhin, Yu.V., Shelekhov, E.V.: Phase transformations in Fe–Ni system at mechanical alloying and consequent annealing of elemental powder mixtures. Physica B: Condensed Matter 299, 236–241 (2001)
Liu, J., Feng, Y., Qiu, T.: Synthesis, characterization, and microwave absorption properties of Fe–40 wt.%Ni alloy prepared by mechanical alloying and annealing. J. Magn. Magn. Mater 323, 3071–3076 (2011)
Guittoum, A., Layadi, A., Bourzami, A., Tafat, H., Souami, N., Boutarfaia, S., Lacour, D.: X-ray diffraction, microstructure, Mössbauer and magnetization studies of nanostructured Fe 50Ni 50 alloy prepared by mechanical alloying. J. Magn. Magn. Mater 320 (2008)
Gheisari, Kh., Shahriari, Sh., Javadpour, S.: Structural evolution and magnetic properties of nanocrystalline 50 Permalloy powders prepared by mechanical alloying. J. Alloys Compd 574, 71–82 (2013)
Lutterotti, L.: MAUD version 2, 33 (2010) http://www.ing.unitn.it/luttero/maud
Rietveld, H.M.: Line profiles of neutron powder-diffraction peaks for structure refinement. Acta. Cryst 22, 151–152 (1967)
Rietveld, H.M.: A profile refinement method for nuclear and magnetic structures. J. Appl. Crystallogr 2, 65-71 (1969)
Lutterotti, L., Scardi, P.J.: Simultaneous structure and size-strain refinement by the Rietveld method. J. Appl. Cryst 23, 246–252 (1990)
Thomson, P., Cox, D.E., Hastings, J.B.: Rietveld refinement of Debye-Scherrer synchrotron x-ray data from A1203. J. Appl. Cryst 20, 79–83 (1987)
Rowles, M.R., Madsen, I.C.: Whole-pattern profile fitting of powder diffraction data collected in parallel-beam flat-plate asymmetric reflection geometry. J. Appl. Cryst 43, 632–634 (2010)
Balzar, D., Popa, N.C.: Analyzing microstructure by Rietveld refinement. Rigaku J 22, 16–25 (2005)
Langford, J., Wilson, A.: Scherrer after sixty years: a survey and some new results in the determination of crystallite size. J. Appl. Cryst 11, 102–113 (1978)
Scherrer, P.: Bestimmung der Grösse und der inneren struktur von Kolloidteilchen Mittels Röntgenstrahlen, nachrichten von der Gesellschaft der Wissenschaften, Göttingen, ”Mathematisch-Physikalische Klasse, 2, 98–100 (1918)
Stokes, A.R., Wilson, A.J.C.: The diffraction of x-rays by distorted crystal aggregates. I. Proc. Phys. Soc. Lond 56, 174–181 (1944)
Mittemeijer, E.J., Welzel, U.: The “state of the art” of the diffraction analysis of crystallite size and lattice strain. Z. Kristallogr 223, 552–560 (2008)
Lutterotti, L., Scardi, P.: Simultaneous structure and size–strain refinement by the Rietveld method. J. Appl. Cryst 23, 246–252 (1990)
Lagarek, K., Rancourt, D.: Recoil Software, Physics Department. University of Ottawa (1998)
Jartych, E., żurawicz, J.K., Oleszak, D., Pekala, M.: X-ray diffraction, magnetization and Mössbauer studies of anocrystalline Fe–Ni alloys prepared by low- and high-energy ball milling. J. Magn. Magn. Mater 208, 221–230 (2000)
Chicinas, I., Pop, V., Isnard, O., Le Breton, J.M., Juraszek, J.: Synthesis and magnetic properties of Ni3Fe intermetallic compound obtained by mechanical alloying. J. Alloys Compd 352, 34–40 (2003)
Hamlati, Z., Guittoum, A., Bergheul, S., Souami, N., Taibi, K., Azzaz, M.: X-ray diffraction, microstructure, and Mossbauer studies of Fe 72Al 28 alloy elaborated by Mechanical milling. J. Mater. Eng. Perform 21 (2012)
Kezrane, M., Guittoum, A., Boukherroub, N., Lamrani, S., Sahraoui, T.: Mössbauer and X-ray diffraction studies of nanostructured Fe 70Al 30 powders elaborated by mechanical alloying. J. Alloys Compd 536S, S304–S307 (2011)
Goddard, W.A., Brenner, D.W.: Handbook of nanoscience engineering and technology, pp 590–630. CRC Press, London (2003)
De Keijser, Th.H., Mittemeijer, E.J.: Correction for errors in microstrain values from X-ray diffraction line profiles. Phil. Mag. A 36, 1261–1264 (1977)
Delhez, R., De Keijser, Th.H., Mittemeijer, E.J. (1980)
Hamzaoui, R., Elkedim, O., Fenineche, N., Gaffet, E., Craven, J.: Structure and magnetic properties of nanocrystalline alloyed, mechanically Fe–10% Ni and Fe–20% Ni. Mat. Sci. Eng: A 360, 299–305 (2003)
Koohkan, R., Sharafi, S., Shokrollahi, H., Janghorban, K.: Preparation of nanocrystalline Fe–Ni powders by mechanical alloying used in soft magnetic composites. J. Magn. Magn. Mater 320, 1089–1094 (2008)
Hamzaoui, R., Elkedim, O., Gaffet, E., Greneche, J.M.: Structure, magnetic and Mossbauer studies of mechanically alloyed Fe-20 wt.% Ni powders. J. Alloys Compd 29, 32–38 (2006)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kezrane, M., Guittoum, A., Hemmous, M. et al. Elaboration, Microstructure, and Magnetic Properties of Nanocrystalline Fe90Ni10 Powders. J Supercond Nov Magn 28, 2473–2481 (2015). https://doi.org/10.1007/s10948-015-3059-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10948-015-3059-9