
Abstract
A three-dimensional porous hydroxyapatite nanocomposite has been synthesized by a simple, less energy consuming and cost effective one-pot method. In this study, gelatin foam has been used as pore forming agent and incorporated in carboxymethyl cellulose-hydroxyapatite system in composite formation stage. A three-dimensional porous polymers-hydroxyapatite nanocomposite has been formed as a final product. The synthesized porous nanocomposite has been thoroughly characterized by different techniques. It was found that the nanocomposite is highly porous with almost 80% porosity, and has multi-scale pores from 2.5 to 900 μm in size. Furthermore, the synthesized porous composite has compressive strength ~ 11.8 ± 1.5 MPa and modulus ~ 0.243 ± 0.031 GPa, in the range of cancellous bone. Moreover, the nanocomposite provides favorable environment to cells for proliferation, high alkaline phosphatase (ALP) activity and extracellular mineralization. In vitro degradation of synthesized nanocomposites was tested in simulated body fluid. Results ascertained that the synthesized porous hydroxyapatite nanocomposite would be a promising scaffold for bone tissue engineering.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The treatment of severely damaged bones with functional bone substitutes is a major concern for clinicians to get successful post-implantation outcomes. In this sense, three-dimensional synthetic bone grafts have received wide attention in orthopedic applications [1]. However, the suitability and efficacy of these bone substitutes in bone tissue engineering mainly rely on their structural features such as the porosity, pore size and strength [2,3,4,5]. Hence, scaffold, a porous three-dimensional structure attracts the attention of many researchers because it helps in guiding and modulating the growth of cells through its porous structure. However, it requires an interconnected porous system with multi scale pores for nutrient flows, vascular ingrowth and bone tissue formation. Moreover, it needs to be biocompatible, biodegradable and bioresorbable [2,3,4,5,6,7]. So, a proper selection of materials is required. To date, several natural polymers such as collagen, alginate, cellulose, chitosan and gelatin are widely used due to their excellent biocompatibility, biodegradability and hydrophilicity [8,9,10]. Alternatively, ceramics basically calcium phosphates-based ceramics are also used because they possess high stiffness, biocompatibility and osteoconductivity [7, 11, 12]. Despite of their excellent properties, the use of such monolithic scaffolds is limited by significant drawbacks. Generally, the application of polymers is restricted due to their poor strength and high degradability, whereas, ceramics are commonly fragile in nature and processability is very poor. In this perspective, composite scaffolds are always a better option to mitigate these types of problems associated with monolithic scaffolds. So, many researchers focused on the fabrication of composite scaffolds for bone tissue engineering [13,14,15,16,17,18,19].
As we know that an ideal scaffold for bone tissue engineering should possess compositional and mechanical properties analogous to natural bone [2, 4,5,6]. To mimic the compositional–mechanical features of natural bone, nowadays, researchers have developed polymer–hydroxyapatite composite scaffolds. Hydroxyapatite is a calcium phosphate [Ca10(PO4)6(OH)2] ceramic, structurally and chemically resemblance to inorganic component of natural bone. When it combines with polymers, it improves the mechanical strength, stability and bone binding ability of polymers [7, 12, 13, 15,16,17, 19-22]. So, researchers have employed different techniques to fabricate porous polymer-hydroxyapatite composite scaffolds such as freeze–drying, 3D printing, electro-spinning and polymer templating etc. [3, 7, 12, 13, 15,16,17,18,19,20,21,22,23]. Most of these techniques are complex, multistep, energy consuming and cost intensive in nature. These days, researchers focus on the synthesis of porous material through simple and cost effective method, like, microfluidic and gyratory methods are two emerging methods [24,25,26]. Gultekinoglu et al. synthesized honey comb like porous structure by microfluidic method [25]. Recently, Mahalingam et al. demonstrated pressurised gyratory method for the fabrication of core-sheath polymeric fibrous structure as scaffold for biomedical applications [26]. Hence, studies show that the structure of material can be modulated with the variation of fabrication process and synthetic method.
In the present study, we also aimed to develop a three-dimensional polymer-hydroxyapatite composite scaffold, by a simple, less energy consuming and cost effective method. For this, we have chosen carboxymethyl cellulose as an organic matrix and gelatin as pore forming agent. Both polymers are biocompatible and biodegradable in nature [12, 13, 19, 20, 27,28,29,30,31,32]. Gelatin has been used as pore forming agent in many other reports, where it was generally eliminated by particle leaching and burn out technique to create porous scaffold [20, 29, 30, 33]. But, gelatin is a derivative of collagen and has Arg-Gly-Asp (RGD) sequence of extracellular matrix, imparts good biological properties in the composite [12, 13, 19, 20, 28]. It was also shown in our previous report where the osteogenic activity of cells was increased with the incorporation of gelatin [27]. In that report, we had used carboxymethyl cellulose–gelatin blend for the synthesis of mechanically strong carboxymethyl cellulose–gelatin–hydroxyapatite nanocomposites for weight bearing orthopedic applications. That nanocomposite has strength (compressive strength ~ 40–86 MPa) in the range of cancellous and cortical bone, but less porous (porosity < 50%) in nature. Although, an interconnected porous structure (porosity > 50%) with adequate mechanical strength are the key requirements for bone tissue engineering [2,3,4,5,6]. So, in this report, we prepared gelatin foam and used it to synthesize porous polymer-hydroxyapatite composite scaffold without any elimination. The whole procedure was completed in a single container at ambient temperature; hence, it is named as one pot method. To date, the use of gelatin foam for the fabrication of implantable hydroxyapatite scaffolds have not been studied. This would be a first study where gelatin foam has been used for the synthesis of three-dimensional porous polymer-hydroxyapatite composite scaffold. The synthesized porous polymer-hydroxyapatite nanocomposite was systematically characterized. It was found that the three-dimensional structure has interconnected multi scale pores combined with adequate mechanical strength. In vitro cells study showed that the synthesized composite is biocompatible and bioactive in nature. It confirms the suitability of synthesized three-dimensional composite scaffold in weight bearing application for bone tissue engineering.
2 Materials and method
2.1 Materials
Gelatin, carboxymethyl cellulose (CMC), calcium nitrate, di-ammonium hydrogen phosphate, ammonia solution and ethanol were procured from Merck, India.
2.2 Synthesis of three-dimensional porous polymer–hydroxyapatite composite scaffold
The synthetic procedure of three-dimensional porous polymer–hydroxyapatite composite (pHA) scaffold is very simple to execute in laboratory. Firstly, 1 g CMC was dissolved in 170 mL water. Afterward, 50 mL aqueous calcium solution (0.99 M) was gradually mixed to CMC solution with continuous stirring. Subsequently, ammonia solution was added to maintain the pH of the mixed solution at 10–11 and aged overnight at room temperature. Next day, gelatin foam was produced by whipping of 10 wt% gelatin solution in overhead mechanical stirrer. In the meantime, 100 mL aqueous phosphate solution (0.56 M) was prepared and mixed in Ca–CMC mixture. Subsequently, 60 mL gelatin foam was added to the mixture and stirred well. The slurry was aged for a week at a room temperature. After aging, slurry was carefully put into an oven for drying at 50 °C. A three dimensional porous hydroxyapatite composite scaffold was obtained. The dried scaffold was immersed in ethanol solution for 2 days to wash out unreacted ions and again dried. For study purpose, a general polymer–hydroxyapatite composite (HA) without adding gelatin foam, was also prepared by the same procedure. In the whole experiment, deionized water has been used.
2.3 Characterization
The synthesized composites were structurally characterized by using X-ray diffractometer [XRD, Bruker (D8 Discover)], Fourier-transform infrared spectrometer [FT-IR, Agilent Cary 660], Field emission-scanning electron microscope (FE-SEM Leo s4302 UK), Transmission electron microscope (TEM, CX Philips JEOL 2100). The percentage of porosity was determined by earlier reported method under vacuum condition [22, 29]. The information of pore size and pores distribution were collected from Mercury intrusion porosimeter (PASCAL 140/440), maximum 80 MPa pressure has been used. The compressive strength of scaffolds (length ~ 16 mm, width ~ 8 mm and height ~ 8 mm) was measured using Hounsfield test equipment (H10KS-0290) and followed by ASTM D695.
2.4 In vitro cyto-compatibility test
For in vitro cyto-compatibility test, preosteoblast MC3T3 cells were cultured in alpha minimum essential medium (αMEM) at 37 °C and placed in 5% CO2 supplied incubator. In the present study, cell survivability and proliferation were measured using 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay. For this, 6 × 104 cells were seeded in 6-well plates. According to ISO 10993-5, porous samples (length ~ 2 mm, width ~ 2 mm and thickness ~ 0.2 mm) extract was prepared and added into seeded cells. On every second day, samples extract was replenished with fresh one and maintained for 1, 4 and 7 days at 37 °C and 5% CO2 supplied incubator. After that, absorbance was recorded using Bio-rad plate reader, USA.
Cells behavior in presence of porous composite (pHA) was observed when the pieces of composite (length ~ 2 mm, width ~ 2 mm and thickness ~ 0.2 mm) were directly cultured with cells for 1, 4 and 7 days. At the day of study, images of live cells were taken by phase contrast microscope (MOTIC AE31). To examine the morphology of cells onto the surface of porous composite, cell cultured samples were fixed with p-formaldehyde for 10 min. Then, the samples were dehydrated with graded ethanol solutions, air-dried overnight, sputtered coated with gold and finally observed under scanning electron microscope (SEM).
2.5 In vitro osteogensis study
The alkaline phosphatase (ALP) is an ectoenzyme, secreted by osteoblasts at the time of new bone tissue formation [27, 28]. In this study, ALP activity of pre-osteoblast MC3T3 cells was quantified according the instruction given in ALP estimation kit [CCK035] of HiMedia. Initially, MC3T3 cells were cultured with synthesized nanocomposites for 2 weeks. After incubation for desired period, ALP assay was performed as per the instruction given in the Kit and the absorbance was measured at 405 nm. At final stage, ALP was normalized with total protein content. Total protein content was estimated by Bradford assay kit (HTBC005), HiMedia. In vitro extracellular mineralization by osteoblast is generally investigated by two extensively studied methods alizarin red (AR) staining and von kossa (VK) staining [27, 28]. In the present study, we have evaluated the extracellular mineralization capability of MC3T3 cells in presence of pHA as per the procedure mentioned in our earlier reports [27, 28].
2.6 In vitro degradability study
The in vitro degradability of HA and pHA nanocomposites was evaluated by SBF (simulated body fluid) treatment [27, 34, 35]. For this, SBF was simply prepared by Kokubo method, having 142.0 mM Na+ ion, 5.0 mM K+ ion, 1.5 mM Mg2+ ion, 2.5 mM Ca2+ ion, 147.8 mM Cl− ion, 4.2 mM HCO3− ion, 1.0 mM HPO42− ion and 0.5 mM SO42− ion. Both HA and pHA nanocomposites (length ~ 6 mm, width ~ 3 mm and thickness ~ 3 mm) were immersed in 20 ml SBF by vacuum infiltration and maintained at 37 °C for 7 days, 14 days and 21 days. After that, nanocomposites were withdrawn from SBF, rinsed with water, dried and weighed.
Degradability of nanocomposites was assessed from weight loss (%), which is calculated by–
here W0 is the weight of nanocomposite before SBF treatment; Wt is the weight of SBF treated nanocomposites at time (t). Surface morphology of SBF-treated HA and pHA nanocomposites at different time periods (7 days, 14 days and 21 days) were collected by FESEM.
2.7 Statistical analysis
Statistical analyses of all quantitative assessments were performed by using GraphPad Prism with Student’s t test; the values are significant at p < 0.05. Data were presented as mean ± standard deviation (n = 3).
3 Results and discussion
In the present study, we aimed to fabricate three-dimensional porous polymers–hydroxyapatite nanocomposite having multiscale pores for bone tissue engineering. The scheme of synthetic procedure is presented in Fig. 1. In this method, gelatin foam has been used as pore forming agent and added into reaction mixture. The reaction mixture was aged for 7 days without any disturbance, and the distinct nature of pHA slurry (with gelatin foam) from HA slurry (without gelatin foam) was observed. In the wet condition, HA slurry was settled down in the beaker; however, the pHA slurry was floating on the surface of water. It depicts the successful entrapment of gelatin foam (Fig. S1, supporting information) in slurry and makes it porous, that can be further confirmed from the cross-sectional view of dried pHA composite.

A scheme of synthetic procedure of three-dimensional polymers–hydroxyapatite nanocomposites (top: without gelatin foam; bottom: with gelatin foam)
As we know that the potentiality of any composite is mainly reliant on the advancement of processing route. In composite, processing governs the structure; and the structure in terms regulates the properties of the composite [36]. As in our earlier report, we had synthesized mechanically strong polymer-hydroxyapatite nanocomposite with the same materials viz. carboxymethyl cellulose, gelatin and hydroxyapatite nanoparticles [27]. In that study, gelatin acted as matrix materials and formed a stable polymeric matrix with the association of carboxymethyl cellulose. After that, hydroxyapatite nanoparticles were mineralized in that polymeric matrix and formed three dimensional nanocomposite with less porosity (<50%). However, in this study, a highly porous three dimensional nanocomposite has been successfully synthesized by using gelatin but in different form i.e. foam type. It indicates the role of processing which directly governs the structure of the composite. Here, gelatin foam has been used; as a result a porous three dimensional structure is obtained.
3.1 XRD, FTIR and TEM analyses of synthesized nanocomposites
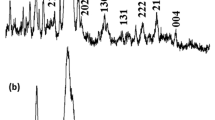
X-ray diffraction (XRD) was carried out to detect the mineral phase of nanocomposites and presented in Fig. 2a. The XRD pattern of HA and pHA nanocomposite showed the diffraction peaks centred at 2θ° ~ 25.89°, 28.9°, 32.09°, 33.81°, 39.81°, 46.96°, 49.63°, 53.35° and 64.13° assigned to (002) (102) (211) (202) (310) (222) (004) (213) and (511) planes of hydroxyapatite, respectively [27, 32]. It also shows that hydroxyapatite is the only mineral phase of the composite and no other secondary phase was found in these XRD patterns. Moreover, the broad nature of XRD peaks depicts the formation of nanocrystalline hydroxyapatite in these composites [27, 31].

a XRD and b FT-IR spectra of synthesized nanocomposites; TEM image of c HA and d pHA nanocomposites
Fourier transform infrared (FT-IR) spectroscopy is another widely used technique to identify the mineral phase of composites. Furthermore, interactions among the components of any composite are also investigated by this technique. FT-IR spectra of HA and pHA nanocomposites are presented in Fig. 2b. In the spectra of both nanocomposites, the typical phosphate bands of hydroxyapatite are found at 1037 cm−1 (P–OH bending), 608 cm−1 (O–P–O bending) and 568 cm−1 (P–O–P stretching) [27, 31]. However, the O–H stretching band of hydroxyapatite and carboxymethyl cellulose overlapped, and gave a broad band at 3422 cm−1 and 3430 cm−1 in HA and pHA nanocomposite, respectively. A distinguishable band at 3165 cm−1 was observed in pHA nanocomposite assigned to N–H group of gelatin. The characteristic anti symmetric band of COO− of carboxymethyl cellulose is present at 1598 cm−1 in HA nanocomposite, whereas this band is shifted to 1649 cm−1 in pHA nanocomposite, which is an indicative of some interactions associated with gelatin [27]. It was further supported by the symmetric band of COO− which is positioned at 1425 cm−1 in HA composite and shifted to 1386 cm−1 in pHA nanocomposite [27].
The shape and size of hydroxyapatite particles in HA and pHA nanocomposites are observed by TEM, and images are presented in Fig. 2c, d. Rod-shaped hydroxyapatite nanoparticles are found in both the composites. Size of hydroxyapatite nanoparticles is same in both the nanocomposites which are almost 20–30 nm length and 10–15 nm wide. It shows that the morphology of hydroxyapatite nanoparticles remains same in the composite after addition of gelatin foam.
3.2 Microstructural analysis of synthesized nanocomposites
The effect of addition of gelatin foam on the microstructure of nanocomposite is observed by FESEM. The microstructure of synthesized nanocomposites is shown in Fig. 3a–f. In FESEM images of HA (Fig. 3a–c), we can see that the composite has polymer layer like structure with small pores, moreover, agglomerated hydroxyapatite nanoparticles were found on this layer. However, pHA composite (Fig. 3d–f) has macro porous structure with pore size ~ 300–900 μm. In Fig. 3e, we have found that the pores are interconnected with ~ 227 μm and 85 μm size strut. This type of interconnected macro porous structure was not found in HA composite (Fig. 3a, b). However, small pores were also found in pHA nanocomposite (Fig. 3f). It shows that pHA nanocomposite has multiscale pores with interconnected porous structure. This interconnected porous structure of pHA composite indicates that the gelatin foam was successfully entrapped in composite formation stage and creates macro/micro porous structure. This interconnected porous structure of composite scaffold would be suitable for accessibility of cells, transportation of nutrients, vascularization and bone tissue ingrowth [2-4].

FESEM images of HA (a–c) and pHA (d–f) nanocomposites
3.3 Pore size distribution and porosity of synthesized nanocomposites
The pore size distribution gives information on how broadly the pore volume is distributed over pores of varying size in the scaffold. In pore size distribution graph (Fig. 4a, b), we can see that the pHA composite has wide range of pores from ~ 2.5 μm to ~ 990 μm in size (Fig. 4b), whereas HA composite (Fig. 4a) has mainly micro pores with average pore diameter ~ 0.02 μm. This observance is in good agreement with microstructural analysis. Furthermore, pHA composite is nearly 80% porous whereas HA composite has almost 10% porosity. The difference can be easily seen in the Fig. 4c, d where HA nanocomposite (Fig. 4c) completely dipped into water whereas pHA composite (Fig. 4d) floats on the surface of water for few hours. However, pHA slowly immersed into water when water completely penetrate into pHA composite, and pHA composite completely dipped in water under vacuum condition. It shows that pHA composite is highly porous with interconnected multi scale pores. The presence of interconnected multi scale pores had already been established to be advantageous for nutrient supply, cell invasion and tissue ingrowth [2,3,4, 7, 8, 12, 14, 21, 37]. A generalization based on many reports has been introduced that the porous scaffolds for bone tissue engineering should possess a porosity greater than 50 vol% and have multi scale pores for multiple applications such as pore diameters < 50 μm facilitates flow of nutrients, physiological liquid exchange and stimulate fibrovascular growth; 50–150 μm pores induce osteoid formation and pores > 150 μm provides enough space for tissue ingrowth and cell colonization [1, 14, 33, 37]. Because of having wide range of pores size, the synthesized three-dimensional porous composite may support all the above-mentioned activities.

Pore size distribution curves of a HA and b pHA nanocomposites; digital images of c HA and d pHA nanocomposite in water
3.4 Mechanical properties
In bone tissue engineering, the mechanical property especially compressive strength is an important criterion in selecting scaffold for load bearing application. In the present study, compressive strength of synthesized composite has been evaluated, and stress vs. strain curves are presented in Fig. 5. HA and pHA composites have compressive strength almost 3.4 MPa and 11.8 MPa respectively. The behaviour of curves is same in both the composites showing viscoelastic deformation. This type of deformation is generally found in human cancellous bone under compression [37]. However, it is known that the mechanical strength of a material generally inverse to its porosity. In contrast, the porous polymer-hydroxyapatite (pHA) composite has higher strength than non/ less porous polymer-hydroxyapatite (HA) composite. This enhancement in mechanical strength of porous composite maybe due to the incorporation of gelatin in carboxymethyl cellulose–hydroxyapatite system, and the existence of intermolecular interactions among CMC, HA and gelatin, improves the strength and modulus of composite [27]. A balance between porosity and mechanical property is a key requirement of any scaffold for clinical weight bearing orthopedic applications.

Compressive stress–strain curve of nanocomposites
The physiomechanical properties of synthesized (HA and pHA) composites and reported CMC-Gel–HA composite were compared with natural bone and summarized in Table 1. It has been found that the pore size, porosity, strength and modulus of pHA composite scaffold are in the range of cancellous bone. However, the compressive strength of reported CMC-Gel–HA composite is in the range of human bone, but the structural features of composite did not match with the natural porous structure of bone which is essential for bone tissue engineering. Moreover, it has been noticed that the compressive strength of CMC-Gel–HA composites is higher (by sevenfold) than the strength of pHA composite. This may be due to less porous (porosity < 50%) structure of CMC-Gel–HA composite and small size of pores (~ 0.01–100 μm). As we know that an interconnected porous three dimensional structure is needed for bone tissue engineering. Because interconnected porous structure provides enough space for bone tissue ingrowth as well as vascularization; and also facilitates space for cell motility. In this sense, it can be said that the compositional–structural–mechanical property of synthesized three-dimensional porous (pHA) composite is suitable for bone tissue engineering, and it is expected that the mechanical property of synthesized pHA nanocomposite, combined with the three-dimensional interconnected porous structure, will significantly improve the performance of bone graft in clinical applications.
3.5 In vitro cyto-compatibility test
In this study, MTT assay was conducted to evaluate the cell survivability and proliferation in presence of porous composite (Fig. 6a). We observed that the absorbance represents the metabolically active live cells was increased with time, in both test sample and control. It shows that the pHA composite is non-toxic in nature and provides suitable environment for cells to behave normally and proliferate with time. The behaviour of cells in presence of pHA composite scaffold is represented in Fig. 6b. In phase contrast images, we observed that the cells are in spindle morphology and the number of cells around the sample was increased with cultured time. It shows the normal activity of cells with good proliferation potential in presence of scaffold. The attachment, growth and proliferation of osteoblast cells on the surface of scaffold are prerequisite for new bone tissue formation. Here, the behavior of MC3T3 pre-osteoblast cells onto the surface of pHA scaffold are shown in Fig. 6b (SEM images). In Day 1, we observed that the MC3T3 cell is present with extensive cytoplasmic extensions spanning the pore walls of the composite. It was mentioned in many reports that the cells divide fast when their cytoskeletons are spread and well organized [6, 7, 14, 18, 21]. This is consistent with our observation that the number of cells was increased on the pHA surface with cultured time and almost covered the porous surface at 7th day. It signifies that pre-osteoblast cells have high affinity for the scaffold surface, that may be due to the presence of HA and gelatin which promotes cell adhesion and proliferation [20, 22, 27]. This was also supported by the large surface area and interconnected porous structure of the composite [21]. It indicates that the surface of scaffold is perfect for cell growth and proliferation.

a MTT assay; b phase contrast and SEM images of cell cultured pHA composite scaffold
3.6 Cell differentiation study
In the present study, cell differentiation was evaluated by measuring the activity of ALP which is an early stage marker of cell differentiation. Differences in ALP activity of cells at different culture times indicate a change in the rate of cell differentiation [27, 28]. ALP activity of cells cultured with pHA nanocomposite was analysed at day 7 and day 14 and the results are shown in Fig. 7a. Cells cultured with pHA nanocomposite showed moderate level of ALP activity at day 7; however, the activity is higher at day 14. As we know that cell differentiation is a follow up phase after cell proliferation [38]. So, the ALP activity of cells is higher in second week. In the final stage of cell differentiation, mineralized bone tissue is formed which is examined by Alizarin red staining (AS) and Von kossa staining (VS) at different time and presented in Fig. 7b. In AS, it was found that calcium ions of newly mineralized bone tissue are stained with red colour [27, 28], and the area of red stained colour was increased with time. In VS, the phosphate groups of mineralized nodules are stained with deep brown colour [27, 28], and the result of VS is consistent with AS. It indicates that three-dimensional porous polymer-hydroxyapatite nanocomposite favours in vitro cell differentiation. It makes it a good choice to be explored as scaffold for bone tissue engineering.

a ALP activity, b Alizarin-red staining (AS) and Von-kossa staining (VS) of MC3T3 cells in presence of pHA nanocomposite
3.7 In vitro degradability study
In this study, the degradation of synthesized nanocomposites was evaluated by calculating the weight loss (%) of samples after SBF treatment. A plot of weight loss (%) of HA and pHA nanocomposites at different time periods is given in Fig. 8a. The weight of both nanocomposites (HA and pHA) are decreased with immersion time, however, the weight loss (%) of pHA nanocomposite is more than HA nanocomposite at each time period. It may be due to porous structure of pHA nanocomposite which provide large surface area to contact with SBF and solubilized polymers more. SEM images of SBF-treated nanocomposites at different time periods are shown in Fig. 8b–g. Comparing the Fig. 8b, c, it can be seen that the number and size of pores on the surface of HA nanocomposite are increased with immersion time. This type of observation is also found in Fig. 8e, f. But, at day 21, the porous structure of composites (HA and pHA) was covered with globular shaped agglomerates, however, the number of agglomerates are larger in pHA nanocomposite (Fig. 8g). Those agglomerates may be newly deposited bone apatite, because this type of deposits is generally found in SBF treated materials [27, 34, 39]. These results showed that both composites were degraded with time and new bone apatite was deposited. However, the porous composite was degraded more with immersion time and large numbers of bone apatite were deposited on the surface of pHA nanocomposite.

a Weight loss (%) of HA and pHA nanocomposites after immersion in SBF for different time periods (0, 7, 14 and 21 days); SEM images of SBF-treated HA (b–d) and pHA (e–g) nanocomposites
4 Conclusions
The present study demonstrated a simple approach to synthesize three-dimensional polymers–hydroxyapatite composite scaffold. The fabrication method by foaming of gelatin creates porous scaffolds with multi-scale pore size and high degree of porosity, which is required for bone tissue engineering. Moreover, the compositional–structural–mechanical property of synthesized composite is analogous to human cancellous bone. The synthesized porous hydroxyapatite composite favours cells to proliferate and differentiate, and also degraded in SBF. It shows that the synthesized porous composite might be a potential candidate as scaffold for bone tissue engineering.
References
A.S. Brydone, D. Meek, S. Maclaine, J. Proc. Inst. Mech. Eng. H 224, 1329–1343 (2010)
D.W. Hutmacher, J.T. Schantz, C.X.F. Lam, K.C. Tan, T.C. Lim, J. Tissue Eng. Regen. Med. 1, 245–260 (2007)
Q.L. Loh, C. Choong, Tissue Eng. B 19, 485–502 (2013)
S. Wu, X. Liu, K.W.K. Yeung, C. Liu, X. Yang, Mater. Sci. Eng. R 80, 1–36 (2014)
Z. Yan, L. Ruixin, F. Yubo, L. Hao, G. Yong, W. Liang, S. Caihong, Z. Dong, Z. Xizheng, J. Porous Mater. 19, 251–259 (2012)
X. Yu, X. Tang, S.V. Gohil, C.T. Laurencin, Adv. Healthc. Mater. 4, 1268–1285 (2015)
J.R. Woodard, A.J. Hilldore, S.K. Lan, C.J. Park, A.W. Morgan, J.A.C. Eurell, S.G. Clark, M.B. Wheeler, R.D. Jamison, A.J.W. Johnson, Biomaterials 28, 45–54 (2007)
M. Jafari, Z. Paknejad, M.R. Rad, S.R. Motamedian, M.J. Eghba, N. Nadjmi, A. Khojasteh, J. Biomed. Mater. Res. B 105, 431–459 (2017)
R. Tylingo, G. Gorczyca, S. Mania, P. Szweda, S. Milewski, React. Funct. Polym. 103, 131–140 (2016)
A. Gloria, R.D. Santis, L. Ambrosio, J. Appl. Biomater. Biomech. 8, 57–67 (2010)
M. Sladkova, M. Palmer, C. Ohman, J. Cheng, S.A. Ansari, M. Saad, H. Engqvist, G.M.D. Peppo, J. Tissue Eng. Regen. Med. 12, 715–726 (2018)
N. Monmaturapoj, W. Soodsawang, W. Thepsuwan, J. Porous Mater. 19, 441–447 (2012)
C. Isikli, V. Hasirci, N. Hasirci, J. Tissue Eng. Regen. Med. 6, 135–143 (2012)
Z. Kang, X. Zhang, Y. Chen, M.Y. Akram, J. Nie, X. Zhu, Mater. Sci. Eng. C 70, 1125–1131 (2017)
R.J. Kane, H.E.W. Bilka, M.J. Meagher, Y. Liu, J.A. Gargac, G.L. Niebur, D.R. Wagner, R.K. Roeder, Acta Biomater. 17, 16–25 (2015)
R.J. Kane, R.K. Roeder, J. Mech. Behav. Biomed. Mater. 7, 41–49 (2012)
F. Sun, H. Zhou, J. Lee, Acta Biomater. 7, 3813–3828 (2011)
R. Cholas, S.K. Padmanabhan, F. Gervaso, G. Udayan, G. Monaco, A. Sannino, A. Licciulli, Mater. Sci. Eng. C 63, 499–505 (2016)
M. Azami, F. Moztarzadeh, M. Tahriri, J. Porous Mater. 17, 313–320 (2010)
S. Fenga, F. Hec, J. Ye, Mater. Sci. Eng. C 82, 217–224 (2018)
J. Zhang, H. Liu, J.X. Ding, J. Wu, X.L. Zhuang, X.S. Chen, J.C. Wang, J.B. Yin, Z.M. Li, ACS Biomater. Sci. Eng. 2, 1471–1482 (2016)
M. Meskinfam, S. Bertoldi, N. Albanese, A. Cerri, M.C. Tanzi, R. Imani, N. Baheiraei, M. Farokhi, S. Farè, Mater. Sci. Eng. C 82, 130–140 (2018)
M.R. Foroughi, S. Karbasi, R.E. Kahrizsangi, J. Porous Mater. 19, 667–675 (2012)
P.L. Heseltine, J. Ahmed, M. Edirisinghe, Macromol. Mater. Eng. 303, 1800218–1800231 (2018)
M. Gultekinoglu, X. Jiang, C. Bayram, K. Ulubayram, M. Edirisinghe, Langmuir 34, 7989–7997 (2018)
S. Mahalingam, S.H. Vanniasinkam, M. Edirisinghe, Mater. Des. 178, 107846–107855 (2019)
C. Sarkar, P. Kumari, K. Anuvrat, S.K. Sahu, J. Chakraborty, S. Garai, J. Mater. Sci. 53, 230–246 (2018)
Z.K. Kuo, P.L. Lai, E.K.W. Toh, C.H. Weng, H.W. Tseng, P.Z. Chang, C.C. Chen, C.M. Cheng, Sci. Rep. 6, 32884–32896 (2016)
M.M. Villa, L. Wang, J. Huang, D.W. Rowe, M. Wei, J. Biomed. Mater. Res. B 104, 1580–1590 (2016)
A. Asif, R. Nazir, T. Riaz, N. Ashraf, S. Zahid, R. Shahid, A.U. Rehman, A.A. Chaudhry, I.U. Rehman, J. Porous Mater. 21, 31–37 (2014)
S. Garai, A. Sinha, Colloids Surf. B 115, 182–190 (2014)
C. Sarkar, A.R. Chowdhuri, A. Kumar, D. Laha, S. Garai, J. Chakraborty, S.K. Sahu, Carbohydr. Polym. 181, 710–718 (2018)
M. Mbarki, P. Sharrock, M. Fiallo, H. ElFeki, Mater. Sci. Eng. C 76, 985–990 (2017)
W. Yang, S.K. Both, Y. Zuo, Z.T. Birgani, P. Habibovic, Y. Li, J.A. Jansen, F. Yang, J. Biomed. Mater. Res. A 103, 2251–2259 (2015)
J. Liuyun, L. Yubao, X. Chengdong, J. Biomed. Sci. 16, 65–75 (2009)
D.D.L. Chung, Mater. Sci. Eng. R 113, 1–29 (2017)
F. Fayyazbakhsh, M.S. Hashjin, A. Keshtkar, M.A. Shokrgozar, M.M. Dehghan, B. Larijani, Mater. Sci. Eng. C 76, 701–714 (2017)
A. Kumar, S. Mandal, S. Barui, R. Vasireddi, U. Gbureck, M. Gelinsky, B. Basu, Mater. Sci. Eng. R 103, 1–39 (2016)
J. Chakraborty, S.D. Sarkar, S. Chatterjee, M.K. Sinha, D. Basu, Colloids Surf. B 66, 295–298 (2008)
Acknowledgements
We would like to thank CSIR-National Metallurgical Laboratory for financial support (OLP-0231) and University Grants Commission for giving fellowship to Chandrani Sarkar.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sarkar, C., Anuvrat, K., Garai, S. et al. One pot method to synthesize three-dimensional porous hydroxyapatite nanocomposite for bone tissue engineering. J Porous Mater 27, 225–235 (2020). https://doi.org/10.1007/s10934-019-00805-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10934-019-00805-y