
Abstract
Mitochondria mediate energy metabolism, apoptosis, and aging, while mitochondrial disruption leads to age-related diseases that include age-related macular degeneration. Descriptions of mitochondrial morphology have been non-systematic and qualitative, due to lack of knowledge on the molecular mechanism of mitochondrial dynamics. The current study analyzed mitochondrial size, shape, and position quantitatively in retinal pigment epithelial cells (RPE) using a systematic computational model to suggest mitochondrial trafficking under oxidative environment. Our previous proteomic study suggested that prohibitin is a mitochondrial decay biomarker in the RPE. The current study examined the prohibitin interactome map using immunoprecipitation data to determine the indirect signaling on cytoskeletal changes and transcriptional regulation by prohibitin. Immunocytochemistry and immunoprecipitation demonstrated that there is a positive correlation between mitochondrial changes and altered filaments as well as prohibitin interactions with kinesin and unknown proteins in the RPE. Specific cytoskeletal and nuclear protein-binding mechanisms may exist to regulate prohibitin-mediated reactions as key elements, including vimentin and p53, to control apoptosis in mitochondria and the nucleus. Prohibitin may regulate mitochondrial trafficking through unknown proteins that include 110 kDa protein with myosin head domain and 88 kDa protein with cadherin repeat domain. Altered cytoskeleton may represent a mitochondrial decay signature in the RPE. The current study suggests that mitochondrial dynamics and cytoskeletal changes are critical for controlling mitochondrial distribution and function. Further, imbalance of retrograde versus anterograde mitochondrial trafficking may initiate the pathogenic reaction in adult-onset neurodegenerative diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Previously, proteomic studies dissected the apoptotic mechanism under oxygen imbalance, showing early signaling molecules that include prohibitin, erythropoietin, RPE65, melatonin, PP2A, and vimentin, exist as anti-apoptotic reagents in the retinal pigment epithelium (RPE) [1–7]. Mitochondrial dysfunction by prohibitin depletion leads to oxidative damage in the RPE [4, 7]. Prohibitin knockdown using small interfering RNA demonstrated that anti-apoptotic function of prohibitin may exist through BCLxL, AIF, BAK, caspase-9 mediated mechanisms. Prohibitin binds to p53, mitochondrial DNA, and phospholipids that include mitochondrial cardiolipin as a multimeric scaffold [4]. Decreased prohibitin leads to apoptotic reactions through mitochondrial disruption, and imbalance of energy metabolism due to decreased mitochondria may accelerate aging and mitochondrial diseases.
The previous models of mitochondrial networks connect the fusion and fission mechanism but not the mitochondrial morphology in detail [8]. We integrated mitochondrial trafficking and morphology for the quantitative analysis of mitochondrial geometry as well as the molecular components of dynamics.
We hypothesized that prohibitin may act as a mitochondria-nucleus shuttle regulating mitochondrial retrograde signaling through prohibitin-cytoskeletal protein complex. The current study aims to understand mitochondrial dynamics and the early apoptotic mechanisms through prohibitin interactions in the RPE. To determine apoptotic pathways in mitochondria and the nucleus, we investigated the prohibitin interactome map using immunoprecipitation data. The prohibitin interactome map showed that a specific cytoskeletal and nuclear protein-binding mechanism may exist to regulate prohibitin signaling as the key element, including vimentin and p53. Our interactome map and immunoprecipitation data further demonstrated that prohibitin may regulate mitochondrial trafficking through two unknown proteins that include 110 kDa protein with myosin head domain and 88 kDa protein with cadherin repeat domain. Cytoskeletal polymers, including microtubules (tubulin), intermediate filaments (vimentin), and microfilaments (actin), were examined using actin, vimentin, and tubulin immunocytochemistry, respectively. To obtain quantitative measurements based on their size, shape, position, and dynamics, we analyzed normal and aberrant mitochondrial networks under oxidative stress as well as cytoskeletal molecules, including actin, tubulin, and vimentin. Our experiments suggest that mitochondria move to the nucleus under stress to fulfill energy demand of the nucleus.
2 Methods
2.1 Protein Interaction
ARPE-19 cells were harvested in a lysis buffer, containing protease inhibitor cocktail, 0.2 % sodium orthovanadate, 10 mM EDTA, 2 mM PMSF, 15 % glycerol, 0.3 % Triton X- 100, 120 mM NaCl in 25 mM Tris. Proteins were collected using sonication (3 × 5 min), centrifugation (13, 000×g, 30 min) after repeated freeze–thaw cycles. Nonspecific binding was minimized using 4 % bead agarose cross-linking. Protein A beads used a buffer containing 7.5 mM NaCl and 0.5 mM sodium phosphate, sodium cyanoborohydride (3 µl, 5 M, 25 °C, 2 h) to immobilized purified prohibitin primary antibody. A buffer containing 5 % glycerol, 1 % NP-40, 0.001 M EDTA, 0.15 M NaCl, and 0.025 M Tris, was used to wash antibody-immbilized protein A column. Prohibitin antibody-protein A column was incubated with crude proteins at 4 °C for overnight. To remove nonspecific binding proteins, column was washed three time using a washing buffer before elution. After elution of three fractions, remaining proteins were mixed using Laemmli sample buffer (2 % SDS, 100 mM DTT). SDS-PAGE separated the elution fractions and visualized Western blot analyzed specific proteins including p53 and prohibitin. Proteins were visualized using Coomassie or silver staining (Bio-Rad, Hercules, CA) and analyzed by mass spectrometry.
All experiments were repeated (n = 3–10 biological samples) with technical duplicate or triplicate. Statistical analysis was performed using StatView software and statistical significance was determined by variance (ANOVA) or unpaired student’s t test when appropriate.
2.2 In Vitro Experiments
ARPE-19 cells were cultured as reported previously [1, 4, 6, 7]. Confluent RPE cells were treated with intense light (7000 lux, 1 h) or tert-butyl hydroperoxide (or H2O2) as oxidative stress (100–200 μM, 24 h) in serum free media. At each time point, cells were harvested and proteins were visualized by immunocytochemistry or Western blotting using primary antibody.
2.3 In Vivo Experiments
NIH guideline and The ARVO (Association for Research in Vision and Ophthalmology) statement on the Use of Animals in Ophthalmic and Vision Research were followed for in vivo experiments. Animals (C3HeB/FeJ, 12 week old female) were obtained commercially (Jackson Laboratory, Bar Harbor). Data from another murine models, including aged and diabetic mice/rats were reported recently [7]. Mice were housed for 2 weeks before the experiments under 250–300 lux of full spectra fluorescent room light, 12 h dark and 12 h light condition. The HALO time represents the hour after light on, meaning light off at HALO 12 and light on at HALO 0. Three groups containing 12 mice/group were used for the current study. The control group was maintained in the 12 h light/12 h dark environment. The constant light group was housed in 250–300 lux for seven days and treated with vehicle solution containing 0.3 % ethanol-saline at HALO 14. The third group was treated with melatonin (50 μg/kg) intraperitoneally under constant light at HALO 14. All mice were euthanized at HALO 18 and the eyes were removed from animals, and retinas were isolated rapidly (microscopic dissection). A buffer containing 10 mM Tris–HCl and 250 mM sucrose removed contaminants and a lysis buffer containing protease inhibitors, 4 % CHAPS, 7 M urea, 2 M thiourea, and 30 mM TrisCl, was used for biochemical analysis.
2.4 Prohibitin Interactome Mapping
Immunocytochemistry and immunoprecipitation identified prohibitin localization and protein interactions. Prohibitin interactome in cancer cells and the retina/RPE were established using protein–protein interaction map software and databases, including STRING 10.0 (http://string-db.org/), iHOP (http://www.ihop-net.org/UniPub/iHOP/), and MIPS (http://mips.helmholtz-muenchen.de/proj/ppi/). Prohibitin interacting proteins [7] were added in STRING query to establish a prohibitin interactome map. Protein interactions were presented using eight categories, including neighborhood (green), gene fusion (red), co-occurrence (dark blue), coexpression (black), binding experiments (purple), databases (blue), textmining (lime), and homology (cyan). Prohibitin interactions were determined and confirmed by genomic context, high-throughput experiments, coexpression, and previous publications in Pubmed.
2.5 Mitochondrial Analysis
Mitochondrial size and morphology were analyzed using the macro connected to Image J software (Ruben Dagda: http://imagejdocu.tudor.lu/doku.php?id=macro:mitpophagy_mitochondrial_morphology_content_lc3_colocalization_macro). We chose the region of interest using the polygon selection tool to analyze mitochondrial morphology. Red, green, and blue individual channels were obtained from the RGB images, then the red and blue channels were closed. The grayscale was extracted from the red channel and the pixels were inverted to photographic channel. The Threshold function determined maximal and minimal pixel values. Cellular area, mitochondrial content, area/perimeter ratio, and mitochondrial circularity were analyzed quantitatively.
2.6 Immunocytochemical Analysis
MitoTracker Orange (100 nM Molecular Probes) was used to analyze mitochondrial morphology. ARPE19 cells were fixed using 10 % formaldehyde (25 min) and the membrane was permeabilized using 0.2 % Triton X-100 (20 min), followed by blocking (0.05 % Tween 20, 10 % FBS, 1 h) and 1:500 anti-prohibitin antibody incubation (4 °C, overnight, antibody from Genemed Synthesis, San Antonio). Prohibitin was visualized using 1:700 Alexa Fluor 488-secondary IgG antibody incubation (25 °C, 1 h, anti-rabbit, Molecular Probes). The nucleus was visualized by DAPI (4, 6-diamidino-2-phenylindole, VECTASHIELD Mounting Medium). A fluorescent microscope was used for image analysis (Zeiss AxioVert 200 M Apo Tome, 63×).
2.7 2D Electrophoresis
ReadyPrep 2-D Cleanup Kit (Bio-Rad, CA) was used for protein purification. BCA protein assay kit was used for protein quantification (Pierce, Rockford, IL). For isoelectric focusing, proteins (150 µg) were mixed with a rehydration buffer (200 µl, 50 mM DTT, 2 % CHAPS, 2 M thiourea, 8 M urea, and 0.5 % DeStreak IPG buffer ranging pH 3–10, GE healthcare, PA) in Immobiline drystrips (11 cm, pH 3–10 and pH 4–7, Bio-Rad, CA) using Ettan IPGphor-3 (GE healthcare). Voltage gradients for 12 kVh was applied (500 V 1 h, 1000 V 1 h, 6000 V 2 h, 8000 V 40 min). Proteins were reduced in an equilibration buffer containing 50 mM DTT, 2 % SDS, 20 % glycerol, 6 M urea and 0.375 M Tris–HCl and alkylated in a buffer containing iodoacetamide (150 mM). Proteins were separated in second dimension using polyacrylimide gels (8–16 %, Bio-Rad, CA) and visualized by Coomassie staining.
2.8 Immunoblotting
Proteins were electrotransferred (30 min, 15 V) onto the PVDF membrane (Bio-Rad), followed by blocking (5 % nonfat dry milk, 0.1 % Tween-20, 4 °C, 2 h). Membranes were incubated in 1:1000 diluted prohibitin primary antibody for overnight at 4 °C (rabbit polyclonal, Genemed Synthesis, San Antonio, TX), followed by a HRP-conjugated secondary antibody incubation at 25 °C for 2 h (1:10,000 dilution, anti-rabbit, Agrisera, Vännäs, Sweden). Phosphorylated vimentin was detected by p-vimentin primary antibody (Santa Cruz Biotechnology CA, sc-16673, 1:500 dilution). Proteins were detected using a chemiluminescent reagent (West Pico, Pierce) and an image analyzer (LAS 4000 mini luminescent, GE, Piscataway, NJ). As a loading control and a positive control, β-actin was analyzed using a mouse monoclonal antibody (1:5000 dilution, Sigma-Aldrich, St. Louis, MO) and a 1:7000 diluted HRP-conjugated secondary antibody (anti-mouse, Santa Cruz Biotechnology, Santa Cruz, CA). Prism and Quantity One software (Bio-Rad) were used for protein quantification based on band area and pixel intensity.
3 Results
3.1 Prohibitin Interactome Mapping
First, we analyzed prohibitin interactions to understand the prohibitin-mediated mitochondrial-nuclear retrograde signaling in the RPE (Fig. 1). Previously , our lipid interaction assay demonstrated that prohibitin is a major cardiolipin-binding chaperone to inhibit the apoptotic signal in mitochondria [4]. Interaction mapping in prohibitin complexes providing direct association was determined using immunoprecipitation, followed by mass spectrometry analysis. Prohibitin binding proteins in the RPE were connected using STRING 10.0 software (http://string-db.org/) [9, 10]. The prohibitin interactome showed an actin and p53 mediated pathways that include direct (physical) and indirect (functional) associations in the map. Then, to compare prohibitin interactome in different cells, we analyzed the prohibitin interactions using prostate/breast cancer cell information (Fig. 2) [11–16]. Prohibitin interactions were confirmed using eight sources that include neighborhood, gene fusion, co-occurence, high-throughput interaction experiments, databases, homology, conserved coexpression, and published knowledge, including ExPASy (http://www.expasy.org/proteomics/protein-protein_interaction), MiPS, and Pubmed database (http://www.ncbi.nlm.nih.gov/pubmed). Our results imply that prohibitin in cancer cells may regulate the transcription through ubiquitin C (UBC), BRE (BRCA complex), E3 ubiquitin ligase (RNF2) and Ras/Raf pathway [11–13, 17]. However, in retinal and RPE cells, activation of prohibitin under stress, including aging, oxidants, light, and insulin depletion, may relay a distinct signaling to CDK2, PI3K, JUN, p53 and actin [18].

Prohibitin Interactome in the RPE. Prohibitin interaction map in retinal (HRPC) and RPE (ARPE-19) cells were determined by immunoprecipitation using a prohibitin antibody. Prohibitin interactome was established using protein–protein interaction map software STRING 10.0 (http://string-db.org/) by adding 40 prohibitin binding proteins in the query [10]. Protein interactions were presented using eight categories, including neighborhood (green), gene fusion (red), co-occurrence (dark blue), coexpression (black), binding experiments (purple), databases (blue), textmining (lime), and homology (cyan). Prohibitin interactions were determined and confirmed by genomic context, high-throughput experiments, coexpression, and previous publications in Pubmed (Color figure online)

Prohibitin Interactome in Cancer Cells. Prohibitin interactome was established using protein–protein interaction map software STRING 10.0 (http://string-db.org/) by adding 40 prohibitin binding proteins in the query [40, 41]. Prohibitin interactome may suggest a new apoptotic signaling or transcriptional regulation based on protein interactions in the retina and RPE. The prohibitin interactome shows functional, indirect associations and direct physical interactions. Prohibitin interactions in cancer cells are derived from eight sources, including neighborhood, gene fusion, co-occurence, high-throughput interaction experiments, conserved co-expression, databases, homology, and published knowledge (Pubmed)
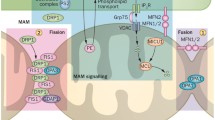
Our prohibitin map showed that specific cytoskeletal and nuclear protein-binding mechanisms may exist to regulate prohibitin signaling as key elements, including actin and BRCA1, to determine the retrograde apoptotic pathway in the RPE. Our immunoprecipitation experiments demonstrated that prohibitin binds to mitochondrial trafficking proteins, including kinesin, unknown protein at 110 kDa (myosin head motor domain, SH3 domain, ATP binding domain, GenBank: BAG 65583, Accession: AK 304840), unknown protein at 88 kDa (cadherin repeat, Ca2+ binding, GenBank: BAC 28880, Accession: AK 034910) (Supplement Table 1).
3.2 Mitochondrial Size Analysis
Next, we analyzed mitochondrial area, circularity, perimeter, content as well as cellular area to determine the correlations between healthy mitochondria and injured mitochondria.
Mitochondrial analysis demonstrated that mitochondrial size decreased and moved to the nucleus under oxidative stress (Fig. 3a). The nucleus, mitochondria, and prohibitin were detected using DAPI, MitoTracker Orange, and the prohibitin-specific primary antibody, respectively (Supplement Figure 1).

Mitochondria (mitochondrial content, morphology, colocalization and mitochondrial autophagy) were analyzed in RPE cells under oxidative stress. Mitochondrial analysis software (Image J with the macro by Dagda RK, U Pitt) was used to calculate mitochondrial morphology quantitatively. The RGB images were split into individual channels (red, green, blue), grayscale conversion, followed by photographic pixel inversion. Quantitative analysis is summarized in Table 1. Nuclear surrounding by mitochondria was shown under stress conditions (a) and extended mitochondrial filament in b (Color figure online)
Previously, our in vitro data using RPE cells demonstrated the positive correlation between apoptotic signaling and mitochondria-nucleus prohibitin shuttling [4]. Mitochondrial morphology regulated by prohibitin and phospholipids, including cardiolipin, may determine RPE cell fate. Mitochondrial analysis software (Image J with the macro by Dagda RK, University of Pittsburgh) was used to calculate mitochondrial concentration quantitatively.
Quantitative analysis that includes mitochondrial content, morphology, and colocalization, was examined in RPE cells under oxidative stress (Fig. 3b). To understand mitochondrial structure and function, twelve categories, including (1) total area of mitochondria, (2) cellular area, (3) mitochondrial content, (4) mitochondrial perimeter, (5) mitochondrial circularity, (6) average perimeter, (7) average mitochondrial area, (8) average circularity, (9) area/perimeter, (10) area/perimeter normalized to minor axis, (11) minor axis, (12) area/perimeter normalized to circularity, were calculated using ARPE-19 cells under oxidative stress (Table 1). Our data showed that mitochondria under oxidative stress change morphology to circular compact shape for fussion, followed by fragmentation toward greater degree of roundness and circularity. Total area of mitochondria decreased in 40–50 % and both perimeter/circular mitochondria were downregulated up to 60–70 %. Area/perimeter normalized to circularity ratio of mitochondria was decreased to 63 % (60 min) under oxidative stress, showing positive correlation between mitochondrial morphology versus apoptotic RPE.
Our data show that RPE mitochondria change their size (smaller network vs larger network), shape (non-tubular, swollen tubes, more branched), connectivity (open vs. closed), position (uniform distribution, asymmetric distribution, close to the nucleus, far from the nucleus) and dynamics (over-fused, over-fragmented) under stress conditions (Fig. 4).

Mitochondrial morphology changes under oxidative stress. Mitochondrial structure in healthy RRE cell was changed in the apoptotic RPE cell. Tubular mitochondria were turned into non-tubular, closed structure
3.3 Mitochondrial Decay and Cytoskeletal Changes
Our unbiased proteomic approach demonstrated that actin, tubulin, and vimentin filaments were changed dramatically in the RPE and retina under oxidative environment [2, 3, 6]. Oxidants disrupt barrier integrity and cell junction to initiate the RPE pathogenesis through a blood-retina barrier degeneration mechanism. Balance between actin filaments, intermediate filaments, and microtubules regulates RPE cytoskeleton, mitochondrial morphology as well as mitochondrial traffic. Vimentin phosphorylations induce disassembly of intermediate filaments in vitro [6, 19]. Melatonin is synthesized nocturnally to protect the retina and a potential therapy for retinal diseases. To determine downstream regulations of the retina protection by melatonin, we designed the experiments using melatonin under constant light. Our 2D SDS-PAGE, Western blotting, and mass spectrometry analysis of vimentin shows site-specific phosphorylations in vivo (Fig. 5). Mice were exposed to three different conditions; (1) 12 h light (250–300 lux)/12 h dark, (2) 24 h continuous light (24 h) for seven days, (3) 24 h continuous light plus melatonin treatment. Twenty-four hours of constant light for seven days upregulated PP2A (box 1) and vimentin (box 2), 1.8 fold and two fold respectively, which were down-regulated by melatonin treatment under 24 h constant light condition (Fig. 5a). Post-translational modifications in cytoskeletal proteins detected by mass spectrometry were listed in Table 2. Vimentin and PP2A showed a reversal of the constant light-induced expression changes after melatonin treatment. Identification of melatonin targets in our study provides a foundation to determine melatonin’s potential to protect mitochondria via vimentin/PP2A mechanism.

Vimentin phosphorylations. 2D SDS-PAGE and Western blot analysis, followed by mass spectrometry shows that vimentin undergoes site-specific phosphorylations and depolymerization under stress conditions. Immunocytochemistry demonstrates stress-induced vimentin aggregation and depolymerization (short filaments). A scaffold subunit of PP2A (#1 box) and vimentin (#2 box) were upregulated under constant light, 1.8 and two fold, respectively (7 days) (a). Target proteins were detected using Coomassie blue staining, Western blotting (b), and analyzed by MALDI-TOF-TOF mass spectrometry (Table 2)
Vimentin is a signal transducing intermediate filament monomer relaying extracellular signal to the nucleus [20–24]. A narrow range of isoelectric focusing was performed for the analysis of vimentin modifications (Fig. 5b). Previously, we presented data from in vivo model from other genetic background (C57BL/6J) under constant light (7 days), intense light (7000–10,000 lux), or circadian regulated (12 h light/12 h dark) as well as C3HeB/FeJ model [1, 3, 33]. Prohibitin signaling and cytoskeletal protein expressions are consistent in mice model with two different genetic background (BL6 vs C3He), however, only albino mice showed different prohibitin/cytoskeletal protein expressions (data not shown). Prohibitin expressions were examined in other murine model (mice vs. rats), in vivo human post mortem tissue, and in vitro model (ARPE19) showing specific prohibitin/cytoskeletal protein expressions under stress conditions [7]. Western blot analysis showed that phosphorylated vimentin tetramer (220 kDa) under normal condition dissociates into phosphorylated monomer (55 kDa) under stress conditions (Fig. 6). Phosphorylated vimentin was visualized by p-vimentin (S55) antibody and comparted to total amount of vimentin as a loading control. Our data suggest that vimentin phosphorylation may determine the net charge (pI), solubility, and depolymerization.

Vimentin depolymerization. Oxidative stress induces accumulation of 57 kDa Ser55 phospho-vimentin and decrease of dephosphorylation of vimentin tetramer. APRE-19 cells were treated with 100, 200 and 500 μM H2O2 for 1 h. Vimentin is used as a loading control. Quantitative analysis shows that vimentin exists as a phosphorylated tetramer (220 kDa) in control, whereas dephosphorylated monomer (55 kDa) or phosphorylated monomer (55 kDa) under stress condition
To determine the interdependence of cytoskeletal assembly, we examined β-tubulin dynamics under stress conditions. Intense light accelerated β-tubulin aggregation as well as localization as shown in Fig. 7. Intense light also leads to actin filament aggregation in cytosol in the RPE as shown in Fig. 8a. Cytosolic β-actin in the dark entered into the nucleus under stress condition. Immunocytochemistry of tubulin and actin demonstrated oxidative stress-mediated aggregation and size changes along with mitochondrial decay (Figs. 7, 8b). Vimentin was shown as an extended filamentous structure in control, however, it was aggregated around the nucleus under stress conditions, including oxidative stress (Fig. 9a) and intense light (7000 lux, Fig. 9b). Cytoskeletal proteins, including actin, tubulin, and vimentin moved into the nucleus under intense light or oxidative stress (Figs. 7, 8, 9).

Cytoskeleton under stress conditions. ARPE19 cells were treated under intense light (7000 lux) for 1 h. Expression changes of cytoskeletal proteins, including tubulin, actin, and vimentin, were visualized by immunocytochemistry. Immunocytochemistry using β-tubulin antibody showed colocalization of tubulin with mitochondria. Western blot analysis shows that neurofilament, vimentin, and tubulin changes their expression levels, whereas actin maintains its concentration under oxidative stress (data not shown)

Immunocytochemical analysis of actin. Control ARPE19 cells were shown in the dark or under stress condition (7000 lux light) (a). Cytosolic actin was observed in the dark, however, actin was translocalized and aggregated outside of mitochondria under intense light. Enlarged (×63) figure represents that β-actin in ARPE-19 cells showed mitochondria-localized actin in control as opposed to nuclear-transported actin in stress conditions (7000 lux light) (b). Aggregation and translocalization were pointed by the white arrow

Intermediate filaments under stress conditions. a RPE cells were incubated under oxidative stress (tert-BuOOH, 200 μM) for 24 h and cells were visualized by immunocytochemistry. b RPE cells were incubated in the dark (control) or intense light (7000 lux). Immunocytochemistry using anti-vimentin antibody, Alexa488 secondary antibody, MitoTracker, and DAPI staining showed vimentin (green) and the nucleus (blue) (Color figure online)
Perimeter/circularity ratio as well as the size of mitochondria were decreased under oxidative stress, showing a positive correlation between mitochondria concentration versus apoptosis (Fig. 10). RPE mitochondria change their size (smaller network vs. larger network), shape (non-tubular, swollen tubes, more branched), connectivity (open vs. closed), position (uniform distribution, asymmetric distribution, close to the nucleus, far from the nucleus) and dynamics (over-fused, over-fragmented) under stress conditions (Fig. 11). When RPE cells were under oxidative stress, mitochondria altered their shape from extended filaments to the small rounded form, possibly by changing microtubule structure by β-tubulin and cardiolipin concentration.

Decreased mitochondria under oxidative environment. RPE cells were incubated using tert-BuOOH or H2O2 (100 μM) for 24 h. For each time point, mitochondria were visualized using MitoTracker Red or Rhodamine123, followed by quantitative analysis using Image J with Mito-Morphology Macro by R. Dagda

Altered mitochondrial morphology under stress. Mitochondrial morphology, content, perimeter, and dynamics were changed under stress conditions. Mitochondrial perimeter/circular ratio as well as total area of mitochondria were downregulated. Mitochondrial size, shape, connectivity, and positions are the determinants of healthy versus apoptotic mitochondria
4 Discussion
How mitochondria move and change their shape remain an intriguing question. The current study tested the hypothesis that mitochondrial trafficking and morphology could correlate positively with the prohibitin complex under stress conditions in the RPE. To obtain quantitative measurements based on mitochondrial size, shape, position, and dynamics, we analyzed normal and aberrant mitochondrial networks under oxidative stress [8]. Mitochondrial membranes may regulate autophagosome through a phospholipids transport mechanism [25]. A protective response mechanism may exist toward extra- and intra-cellular stress under a chronic oxidative environment to activate proliferation reaction in RPE mitochondria [26–28].
Previously, our biochemical studies suggested that mitochondria were disrupted under oxidative stress or RNA knockdown conditions in the RPE through a prohibitin depletion mechanism. The current experiments further test whether an RPE-specific prohibitin interaction network may determine the mitochondrial signaling. Prohibitin interaction networks suggest that prohibitin may regulate mitochondrial traffic and nuclear transcription in the RPE via cytoskeletal proteins and transcriptome-dependent mechanisms.
4.1 Prohibitin Interactome
To determine the major stress responses mediated through mitochondrial protein interactions in the RPE, we established a prohibitin affinity map via integration of immunoprecipitation data.
Integration of protein–protein interaction mapping based on network topology is used to connect prohibitin and potential partners to predict the signaling mechanism. The new algorithm identified the new protein–protein interactions among prohibitin, cytoskeletal proteins, and transcriptional regulators in the nucleus. The current interaction map describes possible prohibitin connections among several pathways that do not appear to be related at first. The network topology identified several potential interactions within each network and between networks, many of which were verified in the literature as well as our immunoprecipitation experiments.
Prohibitin interactions in retinal (HRPC) and RPE (ARPE-19) cells were determined by immunoprecipitation using a prohibitin antibody. Our results imply that prohibitin interactions in cancer cells may regulate transcriptional activation through ubiquitin C (UBC), BRE (BRCA complex), E3 ubiquitin ligase (RNF2) and Ras/Raf pathway. However, in retinal and RPE cells, activation of prohibitin under stress, including aging, oxidants, light, and insulin depletion, may relay a distinct signaling to CDK2, PI3K, JUN, p53 and actin. ARPE19 cell line is one of the most widely used models to study the biology of the RPE, however, limitations exist including the clinical relevance of this in vitro model. Previously, we showed that there is difference of protein expressions between primary RPE cells versus immortalized cells [2]. Prohibitin expressions in vivo from murine model (aged and diabetic rat/mouse) and clinical data (post mortem human AMD eyes) were examined [7].
Decreased cardiolipin in 10–20 % under stress seems to be enough to initiate RPE apoptosis induced by mitochondrial deficits. In the RPE, decreased prohibitin may initiate a cellular protecting mechanism that induces mitochondrial overproliferation.
4.2 Mitochondrial Decay and Prohibitin
In vivo data from human tissue revealed that prohibitin is downregulated in the periphearl region of AMD eyes, suggesting mitochondrial dysfunction and RPE cell death in AMD mechanism [7]. RPE apoptosis is initiated by disintegration of long mitochondrial filaments into small rounded mitochondria moving toward the nucleus. Our data implies that mitochondrial shape, altered cytoskeleton, and prohibitin translocalization are the initial reactions of apoptotic RPE cells [4, 29–31].
Our study showed that a small isolated particle or an extended filament may demonstrate mitochondrial response under oxidative stress. Elongated mitochondrial network may imply a functional electrical charge unit using a phosphate that is required for energy demand. Further, mitochondria organize anti-apoptotic defense system of the RPE when high concentration of O2 is consumed by intense (>7000 lux) or constant light (>24 h). Extracellular stress accelerates proton potential transmission to mitochondria to respond the urgent ATP requirement. A small particle in mitochondria may represent a fast transportable energy complex in the RPE [32, 33].
4.3 Mitochondrial Decay under Oxidative Stress
Our data suggest that mitochondria may indicate cell fate and apoptosis based on size, shape, position, and dynamics. RPE mitochondria change their size (smaller network vs. larger network), shape (non-tubular, swollen tubes, more branched), connectivity (open vs. closed), position (uniform distribution, asymmetric distribution, close to the nucleus, far from the nucleus) and dynamics (over-fused, over-fragmented), responding extracellular environment. When RPE cells were under oxidative stress, mitochondria altered their shape from extended filaments to the small rounded form, possibly by changing microtubule structure and cytoskeletal interactions.
What is the origin of mitochondrial decay and morphological changes? Our previous experiments suggest that decreased cardiolipin and increased cholesterol may determine the apoptotic lipid composition in the RPE. Further, the changes of double bond unsaturation by oxidants may regulate the shape and size of the organelle membrane. Cytoskeletal polymers are essential for mitochondrial dynamics [34, 35]. The cytoskeleton communicates with mitochondria to regulate ATP concentration while their interactions could be connected to various cytoskeletal proteins directly with VDAC (voltage-dependent anion channels) through intermediate filament related proteins.
4.4 Altered Cytoskeleton may Lead to Mitochondrial Decay
Prohibitin-actin interaction led us to examine actin, tubulin, and vimentin filaments in the RPE under stress conditions. Vimentin is involed in light-induced cytoskeleton changes in the retina and the RPE through PP2A and nitric oxide mechanism [6]. N-terminus of vimentin molecule is responsible for intermediate filament interaction with mitochondria. Vimentin-mitochondria interaction decreases mitochondrial motility and elevates mitochondrial membrane potential. Vimentin phosphorylation may determine the net charge (pI), solubility, and depolymerization while immunocytochemistry of actin and tubulin demonstrated oxidative stress-mediated aggregation and size changes along with mitochondrial decay. Oxidants disrupt barrier integrity and cell junction to initiate the RPE pathogenesis through a blood-retina barrier degeneration mechanism. Balance between actin filaments, intermediate filaments, and microtubules regulates RPE cytoskeleton and mitochondrial morphology as well as mitochondrial traffic. Vimentin phosphorylations induce disassembly of intermediate filaments in vitro. Western blot analysis showed that vimentin tetramer dissociates into monomer under stress conditions.
4.5 Prohibitin as a Mitochondrial Trafficking Modulator
Prohibitin is involved in both anterograde (nucleus to mitochondria, bovine retina cell) and retrograde signaling (mitochondria to the nucleus, ARPE-19 cells) [4]. Previously, coimmunoprecipitation of prohibitin suggested that a prohibitin-mediated mitochondrial trafficking complex may exist in the motor-based cytoskeletal polymers that include actin microfilament, vimentin intermediate filament, and microtubules.
Our immunoprecipitation experiments demonstrated that prohibitin binds to mitochondrial trafficking proteins, including kinesin, unknown protein at 110 kDa (myosin head motor domain, SH3 domain, ATP binding domain), unknown protein at 88 kDa (cadherin repeat, Ca2+ binding), actin and lamin B2. Prohibitin-kinesin interaction could be involved in anterograde mitochondrial trafficking, whereas prohibitin-lamin B2 binding may have influence on retrograde transport of mitochondria. Prohibitin binding with a new protein that has myosin motor domain may regulate bidirectional transport of mitochondria along actin microfilaments. Prohibitin-myosin family p110 complex, including PI3K, lamin B2, and cadherin repeating p88, may suggest specific localization of mitochondrial trafficking in the ATP and Ca+2 demanding regions.
Abnormal changes of mitochondrial trafficking is a main pathological initiation reaction in adult-onset neurodegenerative diseases [36–39]. Mitochondrial dysfunction, altered dynamics, impaired transport, and turnover perturbation are associated with the pathology of major neurodegenerative disorders, including Alzheimer’s disease. Impaired mitochondrial transport decreases the release of healthy mitochondria to distal processes, and disrupted eliminations of injured mitochondria from synapses may lead to decreased energy and alteration of Ca2+ concentrartion [40, 41].
Prohibitin-myosin binding may regulate bidirectional transport of mitochondria along actin microfilaments. Actin microfilaments mediate transport over short distance whereas microtubules are the track for long distance transport. Since mitochondrial movements on microtubules are reported to be bidirectional, the polarity orientation of tubulin filaments may also be affected by prohibitin-mediated mitochondrial trafficking patterns. Down-regulation of prohibitin in the RPE may initiate a mitochondrial disruption in AMD. Decreased prohibitin may lead to RPE apoptosis, altered lipid metabolism, and transcriptional reactivation to accelerate the neurodegenerative signaling. Preventing prohibitin depletion may provide a critical therapeutic target as effective strategies for the AMD treatment.
Abbreviations
- AMD:
-
Age-related macular degeneration
- DAPI:
-
4′,6-Diamidino-2-phenylindole
- DIGE:
-
Differential gel electrophoresis
- DMEM:
-
Dulbecco’s modified eagle medium
- IP:
-
Immunoprecipitation
- PP2A:
-
Protein phosphatase 2A
- ROS:
-
Reactive oxygen species
- RPE:
-
Retinal pigment epithelium
- SDS-PAGE:
-
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- siRNA:
-
Small interfering RNA
References
Chung H, Lee H, Lamoke F, Hrushesky WJM, Wood PA, Jahng WJ (2009) Neuroprotective role of erythropoietin by antiapoptosis in the retina. J Neurosci Res 87:2365–2374
Arnouk H, Lee H, Zhang R, Chung H, Hunt RC, Jahng WJ (2011) Early biosignature of oxidative stress in the retinal pigment epithelium. J Proteom 74:254–261
Zhang R, Hrushesky WJM, Wood PA, Lee SH, Hunt RC, Jahng WJ (2010) Melatonin reprogrammes proteomic profile in light-exposed retina in vivo. Int J Biol Macromol 47:255–260
Sripathi SR, He W, Atkinson CL, Smith JJ, Liu Z, Elledge BM, Jahng WJ (2011) Mitochondrial-nuclear communication by prohibitin shuttling under oxidative stress. Biochemistry 50:8342–8351
Lee H, Chung H, Lee SH, Jahng WJ (2011) Light-induced phosphorylation of crystallins in the retinal pigment epithelium. Int J Biol Macromol 48:194–201
Sripathi SR, He W, Um JY, Moser T, Dehnbostel S, Kindt K, Goldman J, Frost MC, Jahng WJ (2012) Nitric oxide leads to cytoskeletal reorganization in the retinal pigment epithelium under oxidative stress. Adv Biosci Biotechnol 03:1167–1178
Sripathi SR, Sylvester O, He W, Moser T, Um J-Y, Lamoke F, Ramakrishna W, Bernstein PS, Bartoli M, Jahng WJ (2016) Prohibitin as the molecular binding switch in the retinal pigment epithelium. Protein J 35:1–16
Rafelski SM (2013) Mitochondrial network morphology: building an integrative, geometrical view. BMC Biol 11:71
Vidal M, Cusick ME, Barabási A-L (2011) Interactome networks and human disease. Cell 144:986–998
Cusick ME, Klitgord N, Vidal M, Hill DE (2005) Interactome: gateway into systems biology. Hum Mol Genet 14:R171–R181
Zhu B, Zhai J, Zhu H, Kyprianou N (2010) Prohibitin regulates TGF-beta induced apoptosis as a downstream effector of Smad-dependent and -independent signaling. Prostate 70:17–26
Dong P, Jiang L, Liu J, Wu Z, Guo S, Zhang Z, Zhou F, Liu Z (2013) Induction of paclitaxel resistance by ERα mediated prohibitin mitochondrial-nuclear shuttling. PLoS One 8:e83519
Sievers C, Billig G, Gottschalk K, Rudel T (2010) Prohibitins are required for cancer cell proliferation and adhesion. PLoS One 5:e12735
Zhu B, Fukada K, Zhu H, Kyprianou N (2006) Prohibitin and cofilin are intracellular effectors of transforming growth factor beta signaling in human prostate cancer cells. Cancer Res 66:8640–8647
Gamble SC, Odontiadis M, Waxman J, Westbrook JA, Dunn MJ, Wait R, Lam EW-F, Bevan CL (2004) Androgens target prohibitin to regulate proliferation of prostate cancer cells. Oncogene 23:2996–3004
Dart DA, Spencer-Dene B, Gamble SC, Waxman J, Bevan CL (2009) Manipulating prohibitin levels provides evidence for an in vivo role in androgen regulation of prostate tumours. Endocr Relat Cancer 16:1157–1169
Liu T, Tang H, Lang Y, Liu M, Li X (2009) MicroRNA-27a functions as an oncogene in gastric adenocarcinoma by targeting prohibitin. Cancer Lett 273:233–242
Vessal M, Mishra S, Moulik S, Murphy LJ (2006) Prohibitin attenuates insulin-stimulated glucose and fatty acid oxidation in adipose tissue by inhibition of pyruvate carboxylase. FEBS J 273:568–576
Vilalta PM, Zhang L, Hamm-Alvarez SF (1998) A novel taxol-induced vimentin phosphorylation and stabilization revealed by studies on stable microtubules and vimentin intermediate filaments. J Cell Sci 111(Pt 1):1841–1852
Whipple RA, Balzer EM, Cho EH, Matrone MA, Yoon JR, Martin SS (2008) Vimentin filaments support extension of tubulin-based microtentacles in detached breast tumor cells. Cancer Res 68:5678–5688
Lahat G, Zhu Q-S, Huang K-L, Wang S, Bolshakov S, Liu J, Torres K, Langley RR, Lazar AJ, Hung MC, Lev D (2010) Vimentin is a novel anti-cancer therapeutic target; insights from in vitro and in vivo mice xenograft studies. PLoS One 5:e10105
Minin AA, Moldaver MV (2008) Intermediate vimentin filaments and their role in intracellular organelle distribution. Biochemistry (Mosc) 73:1453–1466
Esue O, Carson AA, Tseng Y, Wirtz D (2006) A direct interaction between actin and vimentin filaments mediated by the tail domain of vimentin. J Biol Chem 281:30393–30399
Turowski P, Myles T, Hemmings BA, Fernandez A, Lamb NJ (1999) Vimentin dephosphorylation by protein phosphatase 2A is modulated by the targeting subunit B55. Mol Biol Cell 10:1997–2015
Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J (2010) Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141:656–667
Malisan F, Testi R (2003) Mitochondrial lipids as apoptosis regulators. Curr Med Chem 10:1573–1580
Shigenaga MK, Hagen TM, Ames BN (1994) Review Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci USA 91:10771–10778
Choi S-Y, Gonzalvez F, Jenkins GM, Slomianny C, Chretien D, Arnoult D, Petit PX, Frohman MA (2007) Cardiolipin deficiency releases cytochrome c from the inner mitochondrial membrane and accelerates stimuli-elicited apoptosis. Cell Death Differ 14:597–606
Mitchell TW, Buffenstein R, Hulbert AJ (2007) Membrane phospholipid composition may contribute to exceptional longevity of the naked mole-rat (Heterocephalus glaber): a comparative study using shotgun lipidomics. Exp Gerontol 42:1053–1062
Frederick RL, Shaw JM (2007) Moving mitochondria: establishing distribution of an essential organelle. Traffic 8:1668–1675
Sukhorukov VM, Meyer-Hermann M (2015) Structural heterogeneity of mitochondria induced by the microtubule cytoskeleton. Sci Rep 5:13924
Hollenbeck PJ, Saxton WM (2005) The axonal transport of mitochondria. J Cell Sci 118:5411–5419
Lee H, Arnouk H, Sripathi S, Chen P, Zhang R, Bartoli M, Hunt RC, Hrushesky WJM, Chung H, Lee SH, Jahng WJ (2010) Prohibitin as an oxidative stress biomarker in the eye. Int J Biol Macromol 47:685–690
Rintoul GL, Reynolds IJ (2010) Mitochondrial trafficking and morphology in neuronal injury. Biochim et Biophys Acta Mol Basis Dis 1802:143–150
Sheng Z-H (2014) Mitochondrial trafficking and anchoring in neurons: new insight and implications. J Cell Biol 204:1087–1098
Schwarz TL (2013) Mitochondrial trafficking in neurons. Cold Spring Harb Perspect Biol 5:a011304
Chang DTW, Honick AS, Reynolds IJ (2006) Mitochondrial trafficking to synapses in cultured primary cortical neurons. J Neurosci 26:7035–7045
Hopper RK, Carroll S, Aponte AM, Johnson DT, French S, Shen R-F, Witzmann FA, Harris RA, Balaban RS (2006) Mitochondrial matrix phosphoproteome: effect of extra mitochondrial calcium. Biochemistry 45:2524–2536
Schlager MA, Hoogenraad CC (2009) Basic mechanisms for recognition and transport of synaptic cargos. Mol Brain 2:25
Wang S, Fusaro G, Padmanabhan J, Chellappan SP (2002) Prohibitin co-localizes with Rb in the nucleus and recruits N-CoR and HDAC1 for transcriptional repression. Oncogene 21:8388–8396
Thuaud F, Ribeiro N, Nebigil CG, Désaubry L (2013) Prohibitin ligands in cell death and survival: mode of action and therapeutic potential. Chem Biol 20:316–331
Acknowledgments
We thank Dr. Harold J. Sheedlo (University of North Texas Health Science Center) for providing HRP cells. We thank Dr. Cameron Atkinson, Trevor Moser, Hyunju Lee, Dr. Hilal Arnouk, Dr. Ruonan Zhang, and Alex Keim for their excellent technical assistance. This study was supported by the Century II Equipment Fund, Research Excellence Fund from Michigan Technological University, Equipment Fund from NSF, Research and Teaching assistantship from American University of Nigeria. This work was supported in part by an unrestricted grant from Research to Prevent Blindness to the University of Utah Department of Ophthalmology and Visual Sciences. The authors thank Dr. Tristan Purvis for his critical reading and suggestions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Figure 1
Decreased mitochondria and prohibitin translocalization under oxygen imbalance conditions. Proteins in RPE cells treated by H2O2 (100 μM) were detected by immunocytochemistry. MitoTracker Orange, DAPI, prohibitin-specific primary antibody and Alexa488-conjugated secondary antibody showed mitochondria, the nucleus and prohibitin, respectively (PDF 704 kb)
Table 1
Immunoprecipitation experiments demonstrated that prohibitin binds to mitochondrial trafficking proteins, including kinesin, unknown protein at 110 kDa (myosin head motor domain, SH3 domain, ATP binding domain, GenBank: BAG 65583, Accession: AK 304840), unknown protein at 88 kDa (cadherin repeat, Ca2+ binding, GenBank: BAC 28880, Accession: AK 034910) (DOCX 15 kb)
Rights and permissions
About this article

Cite this article
Sripathi, S.R., He, W., Sylvester, O. et al. Altered Cytoskeleton as a Mitochondrial Decay Signature in the Retinal Pigment Epithelium. Protein J 35, 179–192 (2016). https://doi.org/10.1007/s10930-016-9659-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10930-016-9659-9