
Abstract
Bio-based poly(butylene succinate-co-dodecylene succinate) (PBDSs) were successfully synthesized by adjusting the content of 1,12-dodecanediol (DD) using melt polycondensation method. The chemical structure, thermal behavior, crystallization properties, mechanical properties, and rheological properties were characterized. FTIR, 1 H NMR, and 13 C NMR showed the successful synthesis of polymers with the expected structure. The synthesized polymers have high number-average molecular weight (32500–64200 g/mol). The copolyesters were random copolymers. The relationship between melting temperatures and composition was basically V-shaped distribution, which is a typical characteristic of isodimorphism behavior. Thermogravimetric analysis (TGA) showed that the thermal stability of the synthesized copolyesters was good, with no significant weight loss until 320 °C. The synthesized polymers were all semi-crystalline polymers. When the amount of dodecylene succinate unit in the product is 80%, the copolyester has good mechanical properties (tensile strength: 34.5 MPa, elongation at break: 3489%) and processing properties, so it may be more suitable for making films.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Owing of the economy’s fast growth, petroleum have now become an indispensable resource for people’s daily life and production. Due to the excessive exploitation of petroleum, it is now facing resource depletion. At the same time, non-degradable materials have brought serious environmental pollution and other problems. Therefore, more and more research focuses on the development of materials derived from biomass [1,2,3,4,5,6,7]. Poly(butylene terephthalate) (PBT), poly(propylene terephthalate) (PTT), and poly(ethylene terephthalate) (PET), are mainly synthesized from petroleum. However, bio-based polyesters can be produced from biomass (starch, polysaccharides, or stover) [8,9,10], such as poly(lactic acid) (PLA) [11,12,13], poly(hydroxyalkanoate) (PHA) [14,15,16], and poly(butylene succinate) (PBS) [17,18,19]. Among them, polylactic acid (PLA) can be derived from starch and has high tensile strength and low elongation at break [20]. Additionally, poly(butylene succinate) (PBS) has been applied in large quantities due to its excellent processing properties, and relatively low price [21,22,23]. The prospect of PBS is considerable because PBS is renewable and biodegradable [24,25,26]. However, PBS has poor toughness due to its high crystallinity and large spherical crystals.
Copolymerization is often used to improve the toughness of PBS. Poly(butylene succinate-co-methyl propylene succinate)s (PBMS) were synthesized with 2-methyl-1,3-propanediol (MPO) as the commoner [27]. The crystallinity dropped from 45.12 to 38.2%, and the size of the spherules also shrank. The shift from brittle fracture to ductile fracture was realized by the rise in PBMS’s impact strength from 3.9 to 24.6 kJ/m2 and the elongation at break from 10.5 to 71.2%. Shuyi Wu [28] synthesized poly[(butylene succinate)-co-poly(tetramethylene glycol)]s (PBSTMG) with various poly(tetramethylene glycol) (PTMG) contents in order to increase the toughness of PBS. High PTMG content caused the spherulite size to shrink, and increased tear strength. The copolymer’s elongation at break and impact strength have significantly increased due to the phase separation structure and decreased crystallinity. The impact strength of the copolymer with the addition of 10 mol% PTMG increased to 4.5 times that of PBS. The biodegradability of the copolymer was considerably enhanced by the addition of soft segments. Yue Ding [29] synthesized poly(butylene succinate-co-diethylene succinate) (PBSD). The copolymer’s elongation at break was higher than 700%, and its tensile strength is 15 MPa. Yan Zhang [30] synthesized poly(butylene succinate-co-10-butylene hydroxydecanoate)s (PBHs) from 1,4-butanediol (BDO), 1,4-suberic acid (SA), and 10-hydroxydecanoic acid (HDA). PBHs had molecular weights between 24,947 and 45,921 g/mol. With an increase of HDA, PBHs’ melting temperature, crystallization temperature, and Young’s modulus dropped, but their thermal stability and toughness dramatically increased. In addition, aromatic monomers were also used to improve the properties of PBS. High elongation at break (660%) was obtained due to the decrease in crystallinity when the amount of FDCA was 40%.
Since the diols with long chain contain long methylene chains, they can promote increased molecular mobility, which leads to lower glass transition temperatures, improved crystallization kinetics and ductility [31, 32]. 1,12-Dodecanediol (DD) is a kind of renewable diol that can be obtained by whole-cell biotransformation in E. coli [33]. 1,12-Dodecanediol has many broad applications, it can be used as lubricant and surfactant, advanced coatings, plasticizers, and suspending agents, and it can replace 1,6-hexanediol (HDO), while it can provide greater resistance and less water absorption when used in the synthesis of polyesters or polyurethanes [34, 35].
Compared with the diols with short chain, 1,12-Dodecanediol (DD) has 12 methylene groups, which is more suitable for improving the toughness of PBS. In this work, poly(butylene succinate-co-dodecylene succinate) (PBDSs) with different 1,12-dodecanediol contents were synthesized from 1,4-suberic acid (SA), 1,4-butanediol (BDO) and 1,12-dodecanediol (DD). Second, the raw materials can be derived from biomass. The impact of 1,12-dodecanediol on the microstructure, thermal properties, crystallization kinetics and mechanical properties were characterized.
Experiments
Materials
1,4-Succinic acid (SA, 99%) was purchased from Nanjing Chemlin Chemical Industry Co., Ltd. 1,4-Butanediol (BDO, 99%) and tetrabutyl titanate (TBT, ≥ 99%) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. 1,12-Dodecanediol (DD, > 99%) was purchased from TCI Chemical Industry Development Co., Ltd.
Synthesis of PBS, PDS and PBDSs
PBS, PDS and PBDSs polyesters were successfully synthesized by direct esterification and polycondensation reactions. First of all, SA, TBT and BDO/DD were fed into a reactor with a mechanical stirrer. For PBS and PBDSs, the amount of SA was 0.2 mol and the amount of TBT was 60 µL. The molar ratio of (BDO + DD)/SA is 1.1:1. The molar ratio of DD/(BDO + DD) in feed is summarized in Table 1. An electromagnetic stirrer IKA C-MAG HS 7 is used for heating. The reaction was continued under N2 for 4 h when the temperature was raised to 200 °C. Next, raise the temperature to 260 °C while maintaining vacuum for the polycondensation reaction. 2XZ-2 rotary vane vacuum pump is used for remove the diols. The polycondensation reaction was stopped when the rod climbing phenomenon appeared. The stirring speed was modulated by 30 rpm and the vacuum was maintained. The vacuum pump stopped when the temperature cooled to room temperature to prevent the degradation of the synthesized polyester at the high temperature.
For PDS, the amount of SA was 0.15 mol and the amount of TBT was 45 µL. The molar ratio of diol/SA is 1.1:1. The reaction was performed at 200 °C under N2 for 4 h. However, the polycondensation reaction at 260 °C was slow. In the process of polycondensation reaction, the reactor is heated to 260 °C under vacuum for about 8 h, and then heated to 280 °C to speed up the reaction rate. The polycondensation reaction was stopped when the rod climbing phenomenon appeared. The copolymer was named PBDSφSD and φSD% is the molar percentage of dodecylene succinate units in the copolymer.
Characterization
Gel permeation chromatography (GPC): The molecular weight of the polyesters was determined using size exclusion chromatography with a Waters 1525 HPLC pump and a Waters 2414 refractive index detector at 30 °C. Chloroform is the eluent. Polystyrene is the calibration material. A flow rate is 1 mL/min.
Fourier Transform Infrared Spectroscopy (FTIR): Thermo Fisher IS5 FT-IR spectrometer was utilized to analyze the polyester sheets from 4000 to 400 cm−1 with a resolution of 4 cm−1 and 32 scans per sample.
Nuclear magnetic resonance: 600 MHz Bruker system was utilized to measure 1 H NMR and 13 C NMR, and the samples were solubilized in 0.5 mL CDCl3.
Differential Scanning Calorimetry (DSC): 5–10 mg samples were weighed and tested with a DSC TA Q20 instrument in N2 atmosphere at a speed of 50 ml/min. The cooling scan was used to determine the melt crystallization temperature (Tc) and enthalpy (Hc). PBS, PDS, and PBDSs were heated a second time using DSC to determine their melt temperature (Tm) and melt enthalpy (Hm), cold crystallization temperature (Tcc), and cold crystallization enthalpy (Hcc). A primary ramp-up was performed to heat the temperature from − 80 to 160 °C at a velocity of 10 °C/min, dwell for 3 min, then cool down to − 80 °C at a tempo of 10 °C/min and continue a secondary ramp-up to 160 °C at a rate of 10 °C/min.
Thermogravimetric analysis (TGA): The TGA curves were studied using a TGA TA Q50 instrument under N2 atmosphere. The samples were weighed 3–5 mg and heated from 50 to 600 °C at a rate of 10 °C/min.
Wide-angle X-ray diffractometer (WAXD): XRD spectra were recorded on an Ultima IV 2036E102 spectrometer. The scanning speed is 5° min−1 and the scanning range is 5–60°.
Tensile properties: The microcomputer-controlled electronic universal testing equipment was used to evaluate the tensile characteristics of the test specimens. They were hot pressed into sheets with 1 mm thick plates, and then they were cut into dumbbells (4 mm width) using a cutting knife. The crosshead speed was 50 mm/min. Five specimens were tested for each sample. Table 3 displays the data connected to tensile properties.
Rheological properties: The samples were hot pressed into sheets (25 mm diameter, 1 mm thick). Using a TA ARES-G2 rheometer in oscillation mode in a N2 atmosphere at an angular frequency of 10 rad/s, a strain of 1.25%, and a heating rate of 3 °C min−1. The samples’ rheological characteristics were assessed.
Results and Discussion
Synthesis and Chemical Structures of PBS, PDS and PBDSs
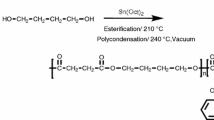
The molecular weights of the synthesized polymers was determined by GPC (Fig. 1). According to Scheme 1, bio-based copolymers were synthesized using a two-step esterification condensation process from BDO, SA, and DD. PBS, PDS and PBDSs had high molecular weights, and the data are listed in Table 1, indicating that the esterification reaction was easily carried out and DD and BDO were able to perform good esterification with SA. Because the by-products are BDO with low boiling point and/or DD with high boiling point, the polycondensation reaction was carried out under evacuation which would draw out the excess diols. DD with high melting temperature was easy to solidify in the distillation tube and blocked the distillation tube, so the distillation tube was heated to help the polycondensation to be completed successfully during the polycondensation reaction.
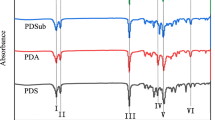
The chemical structures of PBS, PDS, and PBDS were examined by FTIR, 1 H NMR and 13 C NMR. Figure 2 displays the FTIR spectra. Asymmetric and symmetric stretching vibration of C–H in the.

Synthesis of PBS, PDS, and PBDSs from BDO, SA and DD

GPC traces of PBS, PDS and PBDSs

FTIR spectra of PBS, PDS and PBDSs
–CH2− is 2930 and 2850 cm−1, respectively. With an increase in the amount of DD, Peak I (2850 cm−1) steadily rises. The stretching vibration of the carbonyl group is a distinct absorption peak (1755–1670 cm−1). The non-cyclic ester group’s stretching vibration spans the range of 1740–1710 cm− 1, and the peak is at 1710 cm− 1, suggesting that all polyesters exhibit the ester groups. In-plane bending vibration of C–H, the stretching vibration of C–O, and the stretching vibration of C–C are the main vibrations in the range of 1470–1160 cm−1. Bending vibration of methylene appears at 1460 cm−1. The absorption peaks at1210 cm−1 (II) and 1160 cm−1 correspond to the stretching vibration of C–O in butylene succinate unit (BS), and 1260 cm−1 is the stretching vibration of C–O in the ester group. Out-of-plane bending vibration of C–H in the –CH2− is responsible for the weaker absorption peak (920 cm−1). However, an in-plane bending vibration of –CH2−, which is indicative of the existence of long carbon chains, makes up the absorption peak in the III region (725 cm−1). This shows that PBS, PDS, and PBDS were synthesized.
Figure 3 displays the 1 H NMR spectra of PBS, PBDS, and PDS. The results show that there are almost no impurity peaks in the synthetic polyesters. For PBS, the chemical shift of CH2 in the succinate unit was 2.64 ppm (s). The chemical shift of CH2 in the butylene unit was 4.13 ppm (a1) and 1.72 ppm (b1). The chemical shifts of s, a1, and b1 were preserved for PBDSs. Meanwhile, a few more chemical changes emerged: CH2–O–C = O in the DD unit at 4.09 ppm (a2), CH2–CH2–O–C = O at 1.63 ppm (b2), and (CH2)x–O–C = O (where X = 3–6) at 1.28 ppm (d, e, f, g). For PDS, only the chemical shifts (s, a2, b2 and d, e, f, g) were included.
Figure 4 shows 13 C NMR spectra of PBS, PBDSs, and PDS. For PBS, the chemical shift of CH2 in the succinate unit was at 29.02 ppm (t1), the chemical shift of C = O was at 172.29 ppm (c1), and the chemical shifts of CH2 in the butylene unit were at 64.17 ppm (h1) and 25.21 ppm (j1). For PBDSs, the chemical shifts h1 and j1 were retained and the chemical shifts t and c were shifted. Since both BDO and DD are diols with aliphatic chain and they are structurally similar, the shifts of the chemical shifts were small. However, some new chemical shifts appeared: in BSD unit, the chemical shifts of CH2 and C = O of succinate unit near butylene unit were 29.10 ppm (t2) and 172.31 ppm (c2), respectively. The chemical shifts of CH2 and C = O of succinate unit near dodecylene unit were 29.19 ppm (t3) and 172.37 ppm (c3), respectively. In dodecylene unit, the chemical shifts of CH2–O–C = O, CH2–CH2–O–C = O, CH2–CH2–CH2–O–C = O, and (CH2)x–O–C = O (where X = 4–6) is 64.89 ppm (h2), 28.57 ppm (j2), 25.87 ppm (k), and 29.53 ppm (l, m, n), respectively. For PDS, the chemical shifts of h2, j2, k, l, m, and n were retained, and the chemical shifts of CH2 and C = O in succinate unit were shifted to 29.25 ppm (t4) and 172.39 ppm (c4), respectively. Equation (1) were used to calculate molar percentage of DD in the product (ΦDS) from 1 H NMR spectra [36]. The following Eqs. (2, 3, 4) were to determine the molar percentage of DS unit in the product (ΦDS), the average length of the sequences (Ln,BS and Ln,DS), and the randomness of the copolymer (R) according to 13 C NMR spectra [37].

1 H NMR spectra of PBS, PDS and PBDSs

13 C NMR spectra of PBS, PDS and PBDSs (A: the whole spectra, B, C and D: magnification of chemical shifts c, h, t and j)
I a1 and Ia2 are integral intensities of a1 and a2, respectively. The mole ratio of dodecylene succinate unit (DS) is listed in Table 1. It can be seen that the molar ratio of DS in the product is higher than the molar ratio in the feed due to the higher boiling point of DD.
The integrals of h1, h2, c1, c2, c3, and c4 are abbreviated as Ih1, Ih2, Ic1, Ic2, Ic3, and Ic4, respectively. Table 1 provides information about the calculated average sequence lengths and randomness. ΦDS calculated by 1 H NMR and 13 C NMR are nearly identical. As the content of DD increases, Ln,BS decreases and Ln,DS increases. The study also demonstrates that the copolymer’s randomness is close to one, indicating the synthesized copolymers have random structures [38]. Based on the results of FTIR, 1 H NMR and 13 C NMR spectra, PBDSs with the expected chemical structures were successfully synthesized.
Thermal Transition Behaviors and Thermal Stability of PBS, PBDSs and PDS
Figure 5 shows the DSC curves of PBS, PBDSs, and PDS and Table 2 shows the thermal information. The thermal behaviors and melt crystallization behaviors of the copolymers affect the physical properties, processing properties and applications of the copolymers. The thermal behavior of PBDSs depends largely on their components. When the content of dodecylene succinate units in product was less than 50%, Tm of PBDSs decreased with the increase of ΦDS. ΔHm and ΔHc also decreased. When the content of dodecylene succinate units in product was between 57 and 100%, Tm and Tc increased with the increase of ΦDS. As ΦDS increased, the chain regularity of PBDS57, PBDS80 and PDS gradually increased, so their crystallization ability enhanced and the enthalpy increased. PBS shows good crystallization ability during cooling and second heating. In the cooling scan, there was a high enthalpy of melt crystallization at 76.0 °C (ΔHc = 55.0 J/g), and in the second heating scan, there was a high enthalpy of melt at 114.1 °C (ΔHm = 58.3 J/g) and a weak cold crystallization peak at 96.3 °C (ΔHcc = 6.0 J/g). The relationship between Tm (Tc) and composition is basically V-shaped distribution, which is a typical characteristic of isodimorphism behavior. Similar phenomena have been observed in many other copolyesters [39, 40]. When the minor component is completely rejected from the crystalline structure of the major component, the melting temperatures and enthalpies are strongly depressed as the content of the minor component increases in the random copolymer [41].
In the cooling and second heating scan, all polymers showed cold crystallization and melt peaks, indicating that the copolymers have good crystallization ability. In the second heating scan, the copolymers showed significant melt peaks at 83.8 °C (ΔHm = 29.8 J/g), 60.1 °C (ΔHm = 56.2 J/g) and 73.5 °C (ΔHm = 62.3 J/g), respectively. Compared with PBS, PDS also exhibited significant semi-crystalline properties. In the second heating scan, its melt peak and cold crystallization peak appeared at 79 °C (ΔHm = 80 J/g) and 58.3 °C (ΔHc = 75.2 J/g), respectively. However, PDS shows better crystallization ability and crystallization rate than PBS because DD is a long-chain diol and PDS has higher flexibility. PBS can crystallize relatively fast, as evidenced by the fact that the glass transition temperature (Tg) of PBS could not be determined at the second heating scan at 10 °C/min. This is also true for other polymers since the addition of the chains with long CH2 makes the chains more flexible and promotes crystallization more quickly. Only PBS presents Tcc while others do not. This phenomenon has been observed in the DSC curves of PBS in other literature [42]. Compared with PBDS and PDS, PBS contains denser ester bonds and the movement of molecular chains is more difficult due to its shorter BS units, which leads to the possibility that PBS may not crystallize completely during the cooling scan. Therefore, the amorphous region of PBS crystallized and Tcc was observed during the second heating.

DSC curves (A: Cooling, B: Second heating) of PBS, PDS and PBDSs and (C) the relationship between Tm (Tc) and ΦDS in product
For PBS, PDS, and PBDSs, Fig. 6 depicts the TGA and DTG curves in N2. The decomposition temperatures at 5% weight loss (Td,5%) and the highest decomposition rate (Td, max) are listed in Table 2. For PBS, PBDS31, PBDS57 and PBDS80, it’s clear from Fig. 6A that they all show no significant weight loss until 300 °C, indicating that they have very good thermal stability. And it is only when the temperature reaches 350 °C that they undergo significant weight loss. For PDS, it starts to lose weight at around 250 °C because PDS contains long chain diols, so it has the best chain flexibility, leading to its poorer thermal stability than the other four polymers, and also to the lower temperature at which it starts to lose weight. The maximum decomposition temperature of all the synthesised polymers was close to 400 °C, with PBS having a Td,5% and Td,max of 330 and 388 °C, respectively. They were comparable to those found in the literature [43,44,45]. Td,5% and Td,max of PDS were 306 and 407 °C, respectively.

TGA (A) and DTG (B) curves of PBS, PDS and PBDSs under dynamic N2 atmosphere
Isothermal Crystallization Behavior
DSC was used to further examine the isothermal crystallization kinetics of the samples at various isothermal crystallization temperatures (Tc). The temperature was first raised from 25 to 140 °C at a rate of 50 °C/min and maintained for 3 min, and then the temperature was cooled to Tc at a rate of 50 °C/min and maintained for a certain period of time. Finally, the temperature was raised again from Tc to 140 °C at a rate of 10 °C/min. All samples’ crystallization periods tend to lengthen as Tc rises. This suggests that the ability to crystallize is impeded and gradually weakened at higher temperature.
The half-crystallization time (t0.5) was created to compare the variance in isothermal crystallization rate in order to more clearly examine the isothermal crystallization rate [46]. Figure 7 illustrates the connection between t0.5 and Tc. For all samples, t0.5 rises as Tc rises, indicating a slower isothermal crystallization rate at higher Tc. Because the process of the nucleus formation controls the crystallization rate at chosen temperatures and a rise in Tc is unfavorable for the nucleus formation. When t0.5 is close, the crystallization rate is close, so a smaller degree of under cooling (ΔT) indicates better crystallization ability. ΔT is obtained by the following equation.
\({T}_{m}^{o}\) is the equilibrium melting temperature of the sample and Tc is the isothermal crystallization temperature of the sample. ΔT of PBS, PBDS31, PBDS57 and PDS were obtained by the above equation as 33.5, 59.1 34.5 and 14 °C, respectively. Since the crystal structures of PBS and PBDS31, PBDS57 and PDS are similar, PBS has better crystallization ability than PBDS31, while PDS has better crystallization ability than PBDS57.

Isothermal crystallization temperatures (Tc) versus semi-crystallization time (t0.5) for PBS, PDS and PBDSs
Equilibrium Melting Temperature
It is necessary to further investigate the impact of the content of DS unit on the equilibrium melting temperature (\({T}_{m}^{o}\)). After the samples had entirely crystallized at a certain Tc, the Tm values of the samples were acquired by the heating procedure. Figure 8A and B depicts the melting behavior of the PBS and PBDS31 after crystallization and there are two melting peaks. It is clear that as Tc of PBS, PDS and PBDSs rises, the first melting peak shifts toward higher temperature and the area gradually increases. However, the position of the second melting peak nearly stays the same for PBS and PBDS31. The mechanism of melting-recrystallization-remelting may be used to explain this [47, 48]. Figure 8C–E depicts the melting behavior of PBDS57, PBDS80 and PDS after crystallization. They have only one melt peak.

A–E Melting behaviors at 10 °C/min after isothermal crystallization at Tc and (F) the relationship between equilibrium melting temperature and Tc
The Homan–Weeks method is widely used to determine \({T}_{m}^{o}\) [49]. The equilibrium melting temperature in Fig. 8(F) was obtained by the first melting peak, and the intersection of the extension line and the diagonal line was \({T}_{m}^{o}\) because the first melting peak originated from the melting behavior of the original crystals formed during isothermal crystallization. The \({T}_{m}^{o}\) values of PBS, PBDS31, PBDS57, PBDS80, and PDS are 125.5, 114.1, 81.5, 77.2, and 83.0 °C, respectively. The equilibrium melting temperature of PBS is similar to that of literature [45].
Crystal Structures of PBS, PDS and PBDSs
The principles of WAXD and SAXS are basically the same, but WAXD is mainly concerned with crystal structure, crystallinity and molecular chain orientation, while SAXS is mainly concerned with information on the long period of the crystal. In order to measure the crystal structure and crystallinity of polymers, we use WAXD to characterize them. Figure 9 shows the crystal structures of PBS, PDS and PBDSs further obtained by WAXD patterning. First, all the samples were melted at 130 °C and then they were hot pressed at 50 °C for 1 h to obtain crystalline samples.
The obtained PBS, PDS and PBDSs are semi-crystalline polymers. The main diffraction peaks of PBS appear at 19.6°, 21.9° and 22.5°, which can be attributed to the (020), (021) and (110) crystal planes [50, 51]. The weak diffraction peaks of PBS were observed at 25.9° and 28.8°, and these peaks are in agreement with that in references [52,53,54,55]. From the literature, these diffraction peaks are mainly from α-crystals of PBS. The diffraction peaks of PBDS31 are in agreement with those of PBS, proving that PBDS31 also exhibits α-crystals of PBS. However, PBDS57, PBDS80, and PDS have the same diffraction peaks when the content of DS units is greater than 50%, indicating that they have the same crystal structure. It suggests that when the content of DS units rises, the crystal structure alters. The diffraction peak of PBDS57 is stronger than that of PBDS80. PBDS57 has a smaller molecular weight than PBDS80, so the molecular chain of PBDS57 has stronger mobility and is easier to crystallize. The diffraction peak of PDS is the strongest for the following reason: All polymers can crystallize at 50 °C, but PDS is the most crystallizable because PDS has the highest chain flexibility. With the content of BS unit increases, the copolyester’s ability to crystallize is impeded.

WAXD patterns of PBS, PDS and PBDSs
Mechanical Properties
The samples’ tensile strength (σm), yield strength (σy) and elongation at break (εb) were assessed (Table 3). The samples’ stress-strain curves are displayed in Fig. 10A. As illustrated in Fig. 10B, the crystallinity of the tensile samples was examined using WAXD because the mechanical properties are influenced by the crystallinity. The mechanical properties (σm = 37.8 MPa, εb = 672%) of PBS in this study can be compared to that of commercial PBS [56]. Moreover, the elongation at break is significantly higher due to higher molecular weight (Mn = 32,500 g/mol). Compared with tensile strength of PBS, tensile strength of PBDSs dropped as the content of DS unit increased. However, elongation at break of PBDSs was much greater than that of PBS. At the same time, for PBDS31, PBDS57, and PBDS80, tensile strength and elongation at break increased with the content of DS unit increased, which was consistent with the changes of molecular weight and crystallinity. They also showed crisp and similar peaks, and the strength of the diffraction peaks steadily rose as the DD content increased, showing that these materials are more crystalline than the stretched samples, which also show a crystalline condition. PDS has a low elongation at break, which may be due to the formation of large crystals. PBS and PBDS31 displayed similar diffraction peaks in the stretched samples’ WAXD, suggesting that their crystal structures were similar, while PBDS31 was less crystalline than PBS. But PBDS31’s molecular weight is larger than that of PBS and its tensile strength (17.0 MPa) declines dramatically, indicating that tensile strength is mostly impacted by the crystallinity.

A Representative stress–strain curves and B WAXD curves of tensile samples
Rheological Properties
The processing of the polymers is strongly influenced by temperature, and Fig. 11 shows the rheological curves. The material’s viscosity is represented by the loss modulus. Additionally, storage.

Rheological curves of PBS, PDS and PBDSs at different temperatures
modulus evaluates the material’s elastic properties. Complex viscosity, loss modulus, and storage modulus decrease with increasing temperature. Compared with dodecylene succinate unit (DS), butylene succinate unit (BS) is more rigid due to the presence of shorter methylene groups. PBSDS80 with rigid BS and flexible DS is more prone to entanglement than PDS with the flexible DS. Therefore, PBSDS80 have a higher complex viscosity than PDS. The elasticity is waning with increasing temperature, as evidenced by the drop in storage modulus. Storage modulus and loss modulus of the copolyester are higher than those of PBS and PDS. The elastic characteristics of copolyesters are more obvious than those of homopolyesters, which may be due to the fact that the addition of DS units breaks the regularity of molecular chains, making the copolyester more structurally stable and have stronger resistance to deformation. The viscous characteristics of copolyesters are more prominent, likely owing to the fact that the addition of DS units cuts down the flexibility of the molecular chains, resulting in increased resistance to the movement of the chain segments. Due to the better chain flexibility of PDS than PBDS80, the chain segment motion resistance of PBDS80 increases, resulting in a better complex viscosity of PBDS80 than PDS.
Conclusions
We have successfully prepared poly(butylene succinate-co-dodecylene succinate) from DD, SA, and BDO. DD, SA, and BDO are potential bio-based monomers. As the DD content increases, Ln,BS decreases and Ln,DS increases. The copolymers synthesized have random structures. They have high thermal stability. PBS and PBDS31 have the same crystalline structure. However, PBDS57, PBDS80, and PDS have the same crystalline structure. The tensile results demonstrated the copolyesters had good toughness due to lower crystallinity. PBDS80 has superior mechanical properties (tensile strength: 34.5 MPa, elongation at break: 3489%) and it may be more suitable for making films.
Data Availability
The data that support the findings of this study are available on request from the corresponding author upon reasonable request.
References
Sheldon RA (2011) Utilisation of biomass for sustainable fuels and chemicals: molecules, methods and metrics. Catal Today 167(1):3–13
Gallezot P (2012) Conversion of biomass to selected chemical products. Chem Soc Rev 41(4):1538–1558
Iwata T (2015) ChemInform abstract: biodegradable and bio-based polymers: future prospects of eco-friendly plastics. ChemInform 46(18):1
Rabnawaz M, Wyman I, Auras R, Cheng S (2017) A roadmap towards green packaging: the current status and future outlook for polyesters in the packaging industry. Green Chem 19(20):4737–4753
Jeong J, Hussain F, Park S, Kang S-J, Kim J, Stability HT (2020) High tensile strength, and good water barrier property of terpolyester containing biobased monomer for next-generation smart film application: synthesis and characterization. Polymers 12:2458
Hussain F, Park S, Jeong J, Kang S-J, Kim J (2020) Structure–property relationship of poly(cyclohexane 1,4-dimethylene terephthalate) modified with high trans-1,4-cyclohexanedimethanol and 2,6-naphthalene dicarboxylicacid. J Appl Polym Sci 137(32):48950
Hussain F, Jeong J, Park S, Kang S-J, Khan WQ, Kim J (2021) Synthesis and unique characteristics of biobased high tg copolyesters with improved performance properties for flexible electronics and packaging applications. J Ind Eng Chem 100:119–125
Zia KM, Noreen A, Zuber M, Tabasum S, Mujahid M (2016) Recent developments and future prospects on bio-based polyesters derived from renewable resources: a review. Int J Biol Macromol 82:1028–1040
Singhvi M, Gokhale D (2013) Biomass to biodegradable polymer (PLA). RSC Adv 3(33):13558–13568
Zhu Y, Romain C, Williams CK (2016) Sustainable polymers from renewable resources. Nature 540(7633):354–362
Rhim J-W (2013) Preparation and characterization of vacuum sputter silver coated PLA film. LWT Food Sci Technol 54(2):477–484
Castro-Aguirre E, Iñiguez-Franco F, Samsudin H, Fang X, Auras R (2016) Poly(lactic acid)—Mass production, processing, industrial applications, and end of life. Adv Drug Deliv Rev 107:333–366
Chen B-K, Shen C-H, Chen S-C, Chen AF (2010) Ductile PLA modified with methacryloyloxyalkyl isocyanate improves mechanical properties. Polymer 51(21):4667–4672
Kadirvelu K, Kavipriya M, Karthika C, Radhika M, Vennilamani N, Pattabhi S (2003) Utilization of various agricultural wastes for activated carbon preparation and application for the removal of dyes and metal ions from aqueous solutions. Bioresour Technol 87(1):129–132
Zhang J, Shishatskaya EI, Volova TG, da Silva LF, Chen G-Q (2018) Polyhydroxyalkanoates (PHA) for therapeutic applications. Mater Sci Eng: C 86:144–150
Samui AB, Kanai T (2019) Polyhydroxyalkanoates based copolymers. Int J Biol Macromol 140:522–537
Mizuno S, Maeda T, Kanemura C, Hotta A (2015) Biodegradability, reprocessability, and mechanical properties of polybutylene succinate (PBS) photografted by hydrophilic or hydrophobic membranes. Polym Degrad Stab 117:58–65
Tachibana Y, Yamahata M, Kimura S, Kasuya K-i (2018) Synthesis, Physical Properties, and biodegradability of Biobased Poly(butylene succinate-co-butylene oxabicyclate). ACS Sustain Chem Eng 6(8):10806–10814
Tan B, Bi S, Emery K, Sobkowicz MJ (2017) Bio-based poly(butylene succinate-co-hexamethylene succinate) copolyesters with tunable thermal and mechanical properties. Eur Polymer J 86:162–172
Shogren R, Wood D, Orts W, Glenn G (2019) Plant-based materials and transitioning to a circular economy. Sustain Prod Consum 19:194–215
Lee CW, Akashi M, Kimura Y, Masutani K (2017) Synthesis and enzymatic degradability of an aliphatic/aromatic block copolyester: poly(butylene succinate)-multi-poly(butylene terephthalate). Macromol Res 25(1):54–62
Mao H-I, Wang L-Y, Chen C-W, Hsu K-H, Tsai C-H, Cho C-J, Yu Y-Y, Rwei S-P, Kuo C-C (2021) Enhanced crystallization rate of bio-based poly(butylene succinate-co-propylene succinate) copolymers motivated by glycerol. J Polym Res 28(3):92
Jiang D-H, Satoh T, Tung SH, Kuo C-C (2022) Sustainable alternatives to nondegradable medical plastics. Acs Sustain Chem Eng 10(15):4792–4806
Bin T, Qu J-p, Liu L-m, Feng Y-h, Hu S-x, Yin X-c (2011) Non-isothermal crystallization kinetics and dynamic mechanical thermal properties of poly(butylene succinate) composites reinforced with cotton stalk bast fibers. Thermochim acta 525(1):141–149
Tang Y-R, Lin D-W, Gao Y, Xu J, Guo B-H (2014) Prominent nucleating effect of finely dispersed hydroxyl-functional hexagonal boron nitride on biodegradable poly(butylene succinate). Ind Eng Chem Res 53(12):4689–4696
Papageorgiou GZ, Papageorgiou DG, Chrissafis K, Bikiaris D, Will J, Hoppe A, Roether JA, Boccaccini AR (2014) Crystallization and melting behavior of poly(butylene succinate) nanocomposites containing silica-nanotubes and strontium hydroxyapatite nanorods. Ind Eng Chem Res 53(2):678–692
Jiang S, Wang L, Zheng R, Chong Y, Chen Y (2022) Crystallization property, mechanical performance and enzymatic degradation behavior of PBS copolyesters modified by 2-methyl-1,3-propanediol. Polym Eng Sci 62(11):3831–3840
Wu S, Zhang Y, Han J, Xie Z, Xu J, Guo B (2017) Copolymerization with polyether segments improves the mechanical properties of biodegradable polyesters. ACS Omega 2(6):2639–2648
Ding Y, Li S, Wang J, Liu Y, Dong L, Du X, Huang D, Ai T, Ji J (2022) Synthesis, properties, and hydrolysis of bio-based poly(butylene succinate-co-diethylene glycol succinate) copolyesters. J Appl Polym Sci 139(28):e52509
Zhang Y, Yuan W, Liu Y (2017) Synthesis and characterization of bio-based poly(butylene succinate-co-10-hydroxydecanoate). J Elastomers Plast 50(4):325–338
Papageorgiou DG, Guigo N, Tsanaktsis V, Exarhopoulos S, Bikiaris DN, Sbirrazzuoli N, Papageorgiou GZ (2016) Fast crystallization and melting behavior of a long-spaced aliphatic furandicarboxylate biobased polyester, poly(dodecylene 2,5-furanoate). Ind Eng Chem Res 55(18):5315–5326
Perin D, Rigotti D, Fredi G, Papageorgiou GZ, Bikiaris DN, Dorigato A (2021) Innovative bio-based poly(lactic acid)/poly(alkylene furanoate)s fiber blends for sustainable textile applications. J Polym Environ 29(12):3948–3963
Hsieh S-C, Wang J-H, Lai Y-C, Su C-Y, Lee K-T (2018) Production of 1-dodecanol, 1-tetradecanol, and 1,12-dodecanediol through whole-cell biotransformation in Escherichia coli. Appl Environ Microbiol 84(4):e01806–e01817
Youngquist JT, Schumacher MH, Rose JP, Raines TC, Politz MC, Copeland MF, Pfleger BF (2013) Production of medium chain length fatty alcohols from glucose in Escherichia coli. Metab Eng 20:177–186
Griehl W, Ruestem D (1970) Nylon-12-preparation, properties, and applications. Ind Eng Chem 62(3):16–22
Chen X, Chen W, Zhu G, Huang F, Zhang J (2007) Synthesis, 1H-NMR characterization, and biodegradation behavior of aliphatic–aromatic random copolyester. J Appl Polym Sci 104(4):2643–2649
Wang J, Liu X, Zhang Y, Liu F, Zhu J (2016) Modification of poly(ethylene 2,5-furandicarboxylate) with 1,4-cyclohexanedimethylene: influence of composition on mechanical and barrier properties. Polymer 103:1–8
Witt U, Müller R-J, Deckwer W-D (1996) Studies on sequence distribution of aliphatic/aromatic copolyesters by high-resolution 13 C nuclear magnetic resonance spectroscopy for evaluation of biodegradability. Macromol Chem Phys 197(4):1525–1535
Wu T, Wei Z, Ren Y, Yu Y, Leng X, Li Y (2018) Highly branched linear-comb random copolyesters of ε-caprolactone and δ-valerolactone: isodimorphism, mechanical properties and enzymatic degradation behavior. Polym Degrad Stabil 155:173–182
Wang G, Jiang M, Zhang Q, Wang R, Liang Q, Zhou G (2019) New bio-based copolyesters derived from 1,4-butanediol, terephthalic acid and 2,5-thiophenedicarboxylic acid: synthesis, crystallization behavior, thermal and mechanical properties. Polym Test 75:213–219
Pérez-Camargo RA, Arandia I, Safari M, Cavallo D, Lotti N, Soccio M, Müller AJ (2018) Crystallization of isodimorphic aliphatic random copolyesters: pseudo-eutectic behavior and double-crystalline materials. Eur Polymer J 101:233–247
Ding Y, Wang J, Luo C, Yao B, Dong L, Du X, Ji J (2022) Modification of poly(butylene succinate) with biodegradable glycolic acid: significantly improved hydrolysis rate retaining high toughness property. J Appl Polym Sci 139(19):52106
Luo S, Li F, Yu J (2011) The thermal, mechanical and viscoelastic properties of poly(butylene succinate-co-terephthalate) (PBST) copolyesters with high content of BT units. J Polym Res 18(3):393–400
Wu L, Mincheva R, Xu Y, Raquez J-M, Dubois P (2012) High molecular weight poly(butylene succinate-co-butylene furandicarboxylate) copolyesters: from catalyzed polycondensation reaction to thermomechanical properties. Biomacromolecules 13(9):2973–2981
Sun Z, Jiang Z, Qiu Z (2021) Thermal, crystallization and mechanical properties of branched poly(butylene succinate) copolymers with 1,2-decanediol being the comonomer. Polymer 213:123197
Sangeetha VH, Valapa RB, Nayak SK, Varghese TO (2018) Investigation on the influence of EVA content on the mechanical and thermal characteristics of poly(lactic acid) blends. J Polym Environ 26(1):1–14
Liu T, Petermann J (2001) Multiple melting behavior in isothermally cold-crystallized isotactic polystyrene. Polymer 42(15):6453–6461
Qiu Z, Ikehara T, Nishi T (2003) Melting behaviour of poly(butylene succinate) in miscible blends with poly(ethylene oxide). Polymer 44(10):3095–3099
Hoffman JD, Weeks JJ (1962) Melting process and equilibrium melting temperatureof polychlorotrifluoroethylene. J Res Natl Bur Stand Sect Phys Chem 66:13–28
Xu Y, Xu J, Guo B, Xie X (2007) Crystallization kinetics and morphology of biodegradable poly(butylene succinate-co-propylene succinate)s. J Polym Sci Part B: Polym Phys 45(4):420–428
Gan Z, Abe H, Kurokawa H, Doi Y, Microstructures S-S (2001) Thermal properties, and crystallization of biodegradable poly(butylene succinate) (PBS) and its copolyesters. Biomacromolecules 2(2):605–613
Platnieks O, Gaidukovs S, Neibolts N, Barkane A, Gaidukova G, Thakur VK (2020) Poly(butylene succinate) and graphene nanoplatelet–based sustainable functional nanocomposite materials: structure-properties relationship. Mater Today Chem 18:100351
Hu H, Li J, Tian Y, Chen C, Li F, Ying WB, Zhang R, Zhu J (2021) Experimental and theoretical study on glycolic. ACS Sustain Chem Eng 9(10):3850–3859
Liu G, Zheng L, Zhang X, Li C, Jiang S, Wang D (2012) Reversible lamellar thickening induced by crystal transition in poly(butylene succinate). Macromolecules 45(13):5487–5493
Qiu Z, Yang W (2006) Crystallization kinetics and morphology of poly(butylene succinate)/poly(vinyl phenol) blend. Polymer 47(18):6429–6437
Xu J, Guo B-H (2010) Poly(butylene succinate) and its copolymers: research, development and industrialization. Biotechnol J 5(11):1149–1163
Funding
The authors have not disclosed any funding.
Author information
Authors and Affiliations
Contributions
GW: Data curation, Writing-Original draft preparation, Reviewing and editing, Supervision. XH: Data curation, Writing-Original draft preparation, Reviewing and editing. YD: Validation. LZ: Investigation.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wang, G., Hao, X., Dong, Y. et al. Bio-Based Poly(Butylene succinate-co-dodecylene succinate) Derived from 1,12-Dodecanediol: Synthesis and Characterization. J Polym Environ 31, 4990–5002 (2023). https://doi.org/10.1007/s10924-023-02916-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10924-023-02916-9