
Abstract
In the last few years there has been a great deal of interest in modifying the methodologies for obtaining polyurethanes using more sustainable strategies. Following this tendency, in the present work, waterborne UV-curable polyurethanes were synthesized using commercially available dimer fatty acid based polyols obtained from renewable resources (Priplast from Croda). The polyols, characterized by proton nuclear magnetic resonance (1H NMR), differential scanning calorimetry (DSC) and size exclusion chromatography (GPC–SEC), were semicrystalline and presented very broad melting related with their complex molecular weight distribution. Different polyurethane dispersions were obtained by changing the percentage of the hard segment. Comparing with waterborne polyurethane dispersions obtained from non-renewable resources, the dispersions showed higher particle size even using similar or higher amounts of internal emulsifier. The curing behaviour was characterized by photo-differential scanning calorimetry (Photo-DSC) and the results showed that the conversion and polymerization rate decreased with the hard segment content and temperature. The obtained products displayed good thermal characteristics with phase-separated structures.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
It is well known that in the last few decades society has been demanding the development of polymers produced by greener and more sustainable processes [1,2,3]. This is as a result of both the reduction of fossil fuel reserves and also the increase of environmental awareness. It is clear that we are immersed in the transition from the current linear economy to a new concept defined as a circular economy [4, 5]. Currently, the products containing organic solvents and toxic reagents are being restricted by the legislation and as a consequence, there is a great interest not only to develop new green products and processes but also to modify the current methodologies using more sustainable strategies.
Polyurethanes, being one of the polymers most employed in the market, are of course following this tendency [6]. The current research lines to adapt the polyurethane synthesis processes to the market requirements involve mainly the use of renewable feedstock [7,8,9,10], the use of fewer toxic isocyanates [11] or the development of isocyanate free polyurethanes (NIPUs) [12,13,14], the replacement of the tin catalysts [15, 16] and the elimination of organic solvents [17, 18]. All these strategies are introduced to more or less the same extent in the polyurethane market except the development of isocyanate free polyurethanes, which is currently under development.
Although there are some isocyanates based on renewable resources [11], the easier way to introduce green reagents in the polyurethane synthesis is using renewable polyols [19,20,21,22]. All the polyurethane based products are adapting their synthesis processes to the use of renewable feedstock, but the specific characteristics of each product determine the feasibility of replacing fossil fuel based reagents. Thus, renewable polyols, mainly derivated from natural oil are successfully used in the synthesis of polyurethane foams, coatings and adhesives [23,24,25].
In the waterborne polyurethane dispersions (WPUs), the replacement of the conventional polyols is not easy because it has a great effect on the dispersion stability. Thus, Ni et al. [26] synthesized WPU coatings that meet the requirements for industrial application using soybean oil based polyols. Rapeseed oil based polyols were also used to obtain WPUs by Bullermann et al. [27]. Dimer fatty acid based polyester polyols were used by Jiang et al. [28], and by Liu et al. [29] to prepare WPU ureas and by Poussard et al. [7] to prepare WPUs. Finally, Li et al. used isosorbide as chain extender and dimer fatty acid based isocyanate for obtaining renewable WPUs dispersions [30, 31].
Renewable polyols have also been used in the synthesis of UV curable materials [32] but studies related to radiation curable WPUs dispersions are scarcely found. However, these dispersions have a promising future in the development of green polyurethane coatings because they combine the low odour and toxicity of the renewable polyurethane dispersions with the advantages of the UV curing technology. In this way, castor oil and linseed oil based polyols have been used by Mishraab et al. [33] and Chang and Lu [34] to produce UV curable WPUs. Dimer fatty acid based polyols were also used to produce these types of dispersions by Zhang et al. [35].
Recently, our group has studied the UV crosslinking process of radiation curable polyurethane acrylates obtained from non-renewable polyols [36]. The present work is devoted to introducing renewable polyols in these types of systems in order to obtain more sustainable UV curable polyurethane dispersions. Thus, dimer fatty acid based Priplast from Croda was used to replace the petroleum based polyol. Compared with the few studies found in literature regarding these types of systems [35], the present one is the first that determines the curing characteristics of the obtained dispersions by photo-differential scanning calorimetry (Photo-DSC). This data can be of great interest in order to understand the effect of the renewable polyols in the UV curing reaction, with the final objective to develop competitive sustainable UV curable WPUs dispersions.
Experimental Part
Materials
Bio-based polyols (Priplast 3162 (E62) (Mw = 1000 g mol−1) and Priplast 3192 (E92) (Mw = 2000 g mol−1)) were kindly supplied by Croda. Isophorone diisocyanate (IPDI), 1,4-butane diol (BD), 2,2-bis(hydroxymethyl) propionic acid (DMPA), triethylamine (TEA), dibutyltin diacetate (DBTDA), 2-hydroxyethyl methacrylate (HEMA), 1-hydroxycyclohexyl phenyl ketone (Irgacure 184) and acetone were purchased from Sigma Aldrich Chemical Corporation. All materials were used as received.
Synthesis of Polyurethanes
Polymerization reactions were carried out in a 30 mL Schlenk flask reactor under nitrogen atmosphere and magnetic stirring. The reactions were done in three steps. In the first step, polyol, internal emulsifier (DMPA), neutralizing agent (TEA), acetone (70 wt% total solids), the isocyanate (IPDI) and the catalyst (DBTDA, 800 ppm) was added to the reactor. The reaction was carried out at 60 °C until the infrared absorption of the isocyanate group remained constant (about 2 h). In the second step, the chain extender (BD) was added to react with the residual NCO groups (about 1 h). Finally, in the third step, in order to obtain the acrylic end capped prepolymer, HEMA was added and the reaction was carried out until the infrared absorption of the isocyanate group was negligible (about 2 h). Table 1 summarizes the formulations used in the different reactions.
After cooling at room temperature, the reaction mixture was introduced in a flask reactor and the required water amount to obtain a final dispersion containing 35 wt% of solid was added using an addition pump (which is responsible for the water incorporation in a controlled way at 1 mL min−1) at room temperature under a constant mechanical stirring rate of 400 rpm. After water addition, the stirring was kept at the same rate for an additional 30 min. Finally, acetone was removed using a rotary evaporator at 25 °C and at a pressure of 400 mmHg.
UV Curing Process
Dispersions containing 0.3 wt% (to the aqueous waterborne dispersion) of the photoinitiator 1-hydroxycyclohexyl phenyl ketone (Irgacure 184) were prepared dissolving the photoinitiator in a minimum quantity of acetone and adding the solution to the dispersion.
Samples for Photo-DSC were placed in open DSC pans. Water was removed leaving the samples at room temperature overnight in a dark place and afterwards for 2 h in an oven at 80 °C.
Measurements
Fourier transform infrared (FTIR) spectra of bio-based polyols and obtained polymers were recorded onto KBr pellets in a Nicolet 6700 FTIR spectrometer at a resolution of 4 cm−1.
The proton nuclear magnetic resonance (1H NMR) analysis was performed on a Fourier Transform Bruker 300 MHz spectrometer (model Avance 300 DPX) using CDCl3 as solvent.
Size exclusion chromatography (GPC–SEC) was used in order to obtain the molecular weight distribution of polymeric samples. The molecular weight distribution was obtained in a Thermo Scientific Ultimate 300 equipment equipped with an isocratic Dionex UltiMate 3000 pump and a RefractoMax 521 refractive index detector. Tetrahydrofuran (THF) was used as mobile phase at a flow rate of 1 mL min−1. Samples were prepared dissolving the obtained polyurethanes in THF at 2 wt% and filtering using nylon filters with 0.2 µm pore size. Mw, Mn and PI were reported as weight average polystyrene standards.
The particle size (Dp) of the obtained dispersions was determined in a Brookhaven Instruments 90 Plus Particle Size Analyser after adding a drop of the dispersion diluted with distilled water into polystyrene cuvettes.
The Photo-DSC experiments were carried out using a DSC (TA Instruments Q2000) equipped with a photo calorimetric accessory (Omnicure S2000) with a 200 W mercury lamp, giving an optical range from 320 to 500 nm with intensity between 1 and 2 mW cm−2. About 5 mg sample was weighed into aluminium pan and exposed to UV radiation.
The conversion of C=C double bonds (α) was calculated by integrating the area under the exothermic peak using Eq. 1.
where ΔHt is the reaction enthalpy released at time t, and ΔH0theor is the theoretical enthalpy for the complete conversion, that can be calculated using Eq. 2.
where Mwtheor is the molecular weight of the polymer, 54.7 kJ mol−1 is the molar enthalpy of methacrylate group and the functionality is 2 in our case.
The polymerization rate (Rp) was calculated using Eq. 3.
where dα/dt is the polymerization rate, ΔH0theor is the theoretical enthalpy and dH/dt is the measured enthalpy at a constant temperature.
DSC thermograms were recorded in order to determine the glass transition temperature (Tg) and the melting temperature (Tm). The experiments were also carried out using the DSC (TA Instruments Q2000) and were performed between − 80 and 120 °C at a heating rate of 10 °C min−1 under constant N2 flow. Two runs were carried out.
Results and Discussion
Characterization of the Polyols
According to the technical data sheet, Priplast 3162 (E62) and 3192 (E92) are semicrystalline grade difunctional polyols synthesized from Croda Pripol™ C36 dimer fatty acids, a renewable natural resource. Their molecular weights are Mw = 1000 and 2000 g mol−1 respectively.
A deep characterization of the polyols was carried out using FTIR, 1H NMR, GPC–SEC and DSC. FTIR spectrum of E62 polyol measured by transmission is shown in Fig. 1. The main absorptions [3400 cm−1 (OH stretching), 1740 cm−1 (C=O stretching), 1460 cm−1 (CH2 bending), and 1190 cm−1 (C–O–C stretching)] corresponded to an aliphatic ester polyol.

Infrared spectrum of polyol E62
Infrared spectrum of polyol E92 presented the same absorptions but lower intensity of the OH stretching vibration, which is in accordance with the higher molecular weight reported in the data sheet.
Figure 2 shows the 1H NMR spectrum of polyol E62.

1H NMR spectra of E62
According to literature, Priplast polyols are obtained from a mixture of acids (dimer fatty acid and adipic acid) and 1,6-hexanediol [37]. The 1H NMR spectrum shown in Fig. 2 confirmed this structure. The 1H NMR spectrum of polyol E92 showed the same signals but lower relative area of the methylol groups (signal 3′, structure not shown in the figure) that is in accordance with its higher molecular weight. From the areas of the different protons the dimer fatty acid weight percentage was calculated and values of 35 and 33 wt% were obtained for polyol E62 and E92 respectively.
Molecular weight distribution was determined by GPC–SEC. Figure 3 shows the obtained chromatograms.

GPC–SEC chromatograms of samples E62 and E92
The analysis of the chromatograms was performed using polystyrene standards and values of 1779 and 2925 g mol−1 were obtained for the number average molecular weight (Mn) and 1.93 and 2.28 for the polydispersity index for samples E62 and E92 respectively. The values of Mn were higher than the reported in the data sheet (Mw = 1000 and 2000 g mol−1 for samples E62 and E92). This fact was attributed to the calibration with polystyrene.
In addition, it is clear that both samples presented a wide molecular weight distribution containing chains with very different lengths. This is probably due to the high molecular weight of the dimer fatty acid monomer and means that the introduction of only one monomer unit leads to a great increase in the molecular weight. It is important to mention that the molecular weight difference between the peaks corresponding to the lower molecular weight species was close to that of the dimer fatty acid, which confirmed this hypothesis. As a conclusion, the GPC–SEC chromatograms displayed that the dimer fatty acid derived polyols showed a heterogeneous distribution of different molecular weight species in comparison with polyols obtained from non-renewable resources.
Figure 4 shows the DSC thermograms of E62 and E92 polyols.

DSC thermograms of polyols E62 and E92
The samples showed a complex thermal behaviour. At low temperature, both samples showed a glass transition (Tg) around − 60 °C. The value of this transition could not be determined with precision because the equipment did not allow start the run at lower temperatures. Moreover, in the range of − 10 and 42 °C both samples presented a broad endotherm with different maximums that mainly corresponded to the melting of the polyol (Tm). The broadening of the melting transition was a consequence of the existence of species with different molecular weight as showed by the GPC–SEC chromatograms. In addition, the melting temperature of polyol E92 was slightly higher than that obtained in sample E62 due to the lower molecular weight of the latter.
Characterization of the Polyurethanes (PUs)
Samples PU1 and PU2 were prepared with the objective of checking the suitability of the Priplast polyols to react with IPDI. Therefore these samples did not contain DMPA and were not dispersed in water. Both polyols showed good reactivity towards the isocyanate. However, as will be shown in the following sections, after curing, both samples were amorphous. As one of our goals was to obtain semicrystalline materials, the lower molecular weight polyol (1000 g mol−1) was discarded upon the basis that using this polyol it will be more difficult to obtain ordered materials. According to this, water dispersible samples (PU3-6) were prepared using the higher molecular weight polyol (2000 g mol−1). Scheme 1 summarizes the polyurethane synthesis reaction.

Polyurethane synthesis reaction
The obtained systems were characterized employing FTIR and 1H NMR techniques in order to determine the final structure of the synthesized polyurethanes.
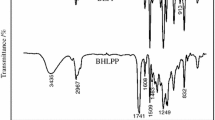
All the samples showed similar infrared spectra. As an example Fig. 5 shows the infrared spectra of sample PU6 obtained both at the beginning and at the end of the reaction.

Initial and final infrared spectra of sample PU6
As can be seen, the relative absorbance of the band centred at 2270 cm−1 attributed to NCO stretching vibrations was present in the initial spectrum but not in the final one, which means that all the isocyanate groups had reacted. At the same time, the absorbance of the bands due to urethane groups increased in the final spectrum (NH stretching band at 3400 cm−1, Amide I at 1720 cm−1 and Amide II at 1550 cm−1). Finally in the spectrum obtained at the end of the reaction, a small band at 1650 cm−1 attributed to the acrylic double bond stretching was observed.
All the polyurethanes were also characterized by 1H NMR spectroscopy to confirm the final structure. All the spectra showed the same signals but with different intensities in the 1H NMR spectrum. Figure 6 shows as an example the spectrum of sample PU6 with the assignation of the main signals.

1H NMR spectrum of sample PU6
The spectrum confirmed the structure of the polymer. Signals at 4.3, 5.6 and 6.1 ppm corroborated the insertion of the acrylic monomer in the polymer structure.
The diameter of the particles (Dp) of the different dispersions was measured and the results are summarized in Table 2.
Sample PU3 gave rise to a non-stable dispersion. It is well known that the increase in the content of carboxylic group (COOH) that could be neutralized by TEA is responsible for the reduction in particle size of aqueous polyurethane dispersions [18]. According to the data shown in Table 2, the minimum amount of DMPA required in order to obtain polyurethane stable dispersion was 0.45 mmol g−1 and high particle size were obtained.
It should be noted that the Dp values were higher than those presented by WPUs containing similar DMPA amounts but obtained from non-bio-based polyols [36]. This result can be explained upon the basis of the lower polarity of the bio-based polyols. It is not easy to compare our data with those reported in literature for polyurethane dispersions obtained from dimer fatty acid based polyols, because in addition to the DMPA amount, factors such as the hard segment content and NCO/OH ratio have a great influence in the particle size. However, it seems that, as in our case, WPUs obtained from these types of polyols [7], or using dimer fatty acid based isocyanates [30], present high particle size. This result can be a drawback for the development of WPUs from these types of polyols.
Photopolymerization Behaviour
In order to study the UV curing process of the dispersions, Photo-DSC measurements were performed and parameters such as conversion (α) and polymerization rate (Rp) were studied.
Figure 7 shows the conversion (α) of the curing reaction as a function of the reaction time for sample PU4 cured at 10 °C and sample PU6 cured at 10 and 30 °C.

Curing conversion of PU4 cured at 10 °C and PU6 cured at 10 and 30 °C
The slope of the conversion curve at the beginning is related to the UV curing rate and the plateau value at the end of curing indicates the final conversion.
It is clear that, regardless of the sample, the final conversion reached by the samples cured at 30 °C was close to 0.6. This fact indicates that the hard segment content of the samples did not affect the final conversion. However, the sample cured at 10 °C showed a significant reduction in the final conversion (conversion of 0.2). The temperature rise improved the segmental mobility of the polymer chains and made the unsaturation sites more accessible for polymerization, thereby increasing the conversion.
Figure 8 shows the polymerization rate (Rp) versus time for PU4 and PU6 samples. There was a clear difference between the two photopolymerization temperatures (10 and 30 °C). In literature it is shown that in general the rate of polymerization increases with temperature, this is also observed in our work. All the curves showed an initial rapid increase in the polymerization rate, meaning that the maximum polymerization rate was reached in the first minutes. The decrease of the polymerization rate at higher conversions was related to the immobilization caused by the curing reaction. In addition, the Rp maximum gradually shifted to longer times as temperature decreased.

Polymerization rate (Rp) of sample PU4 cured at 10 °C and sample PU6 cured at 10 and 30 °C
In addition, for the same curing temperature, PU6 had lower Rpmax than PU4. The higher hard segment percent of sample PU6 could be the responsible for this behaviour considering that the hard segment reduces chain mobility and therefore reduces the diffusion rate of the radicals.
Finally, it is worth mentioning that both the conversion and polymerization rate values reached with polyurethanes synthesized from dimer fatty acid diols were slightly lower in comparison with those reported in literature for non-bio-based polyols [36]. This result could be related with the semicrystalline nature of the dimer fatty acid polyols used in the present work.
Thermal characteristics of the cured samples evaluated by DSC. The obtained thermograms are reported in Fig. 9.

DSC thermograms for all the synthetized samples
As can be seen, all the samples showed a glass transition at low temperatures related to the polyol. In addition to the glass transition, some of the samples presented an endotherm close to 30 °C related to polyol melting.
Compared with pure polyol (Fig. 4), in all cases the glass transition was shifted to higher temperatures in the polyurethanes. This fact was a consequence of the partial mixing between the segments in the polyurethane. In addition, the sample obtained from the lower molecular weight polyol showed a higher Tg, which emphasised that in the lower molecular weight sample the phase separation between the segments was lower [38].
In addition to the molecular weight, the hard segments content is an important factor in determining the thermal properties of the synthesized polyurethanes. In all the samples, Tg shifted to lower temperatures with the increase of hard segment, which means than the samples obtained from high hard segment content were more phase separated.
Finally it is important to note that the final products presented low crystallization capacity. Thus, the sample obtained from the lower molecular weight polyol was not able to crystallize and only the samples containing hard segment content higher than 50% were able to show a semicrystalline behaviour.
Conclusions
Waterborne UV-curable polyurethanes were synthetized using dimer fatty acid based polyols. Compared with WPUs obtained from non-renewable resources the dispersions showed a higher particle size. Moreover, the samples were able to photopolymerize, and the Photo-DSC results showed that the conversion and polymerization rate decreased with the hard segment content and temperature. The obtained products displayed good thermal characteristics with phase separated structures. However, we were not able to obtain dispersions with an appropriate particle size using DMPA amounts similar or higher than those used in literature, which limits the use of these polyols in the development of greener UV curable WPUs.
References
Cheng HN, Gross NA, Smith PB (2015) ACS symposium series. American Chemical Society, Washington DC
Gandini A, Belgacem MN (2002) J Polym Environ 10:105
Gandini A (2008) Macromolecules 41:9491
Pomponi F, Moncaster A (2017) J Clean Prod 143:710
Winans K, Kendall A, Deng H (2017) Renew Sustain Energy Rev 68:825
Noreen A, Zia KM, Zuber M, Tabasum S, Zahoor AF (2016) Prog Org Coat 91:25
Poussard L, Lazko J, Mariage J, Raquez JM, Dubois P (2016) Prog Org Coat 97:175
Sakulsaknimitr W, Wirasate S, Pipatpanyanugoon K, Atorngitjawat P (2015) J Polym Environ 23:216
Cateto CA, Barreiro MF, Rodrigues AE, Belgacem MN (2011) React Funct Polym 71:863
Gurunathan T, Mohanty S, Nayak SK (2015) Prog Org Coat 80:39
Calvo-Correas T, Santamaria-Echart A, Saralegi A, Martin L, Valea A, Corcuera MA, Eceiza A (2015) Eur Polym J 70:173
Cornille A, Auvergne R, Figovsky O, Boutevin B, Caillol S (2017) Eur Polym J 87:535
Rix E, Grau E, Chollet G, Cramail H (2016) Eur Polym J 84:863
Maisonneuve L, Lamarzelle O, Rix E, Grau E, Cramail H (2015) Chem Rev 115:12407
Sardon H, Pascual A, Mecerreyes D, Taton D, Cramail H, Hedrick JL (2015) Macromolecules 48:3153
Sardon H, Irusta L, Fernández-Berridi MJ (2009) Prog Org Coat 66:291
Nanda AK, Wicks DA, Madbouly SA, Otaigbe JU (2005) J Appl Polym Sci 98:2514
Sardon H, Irusta L, Fernández-Berridi MJ, Luna J, Lansalot M, Bourgeat-Lami E (2011) J Appl Polym Sci 120:2054
Gandini A, Lacerda TM (2015) Prog Polym Sci 48:1
Lligadas G, Ronda JC, Galiá M, Cádiz V (2013) Mater Today 16:337
Kathalewar M, Sabnis A, D’Melo D (2014) Prog Org Coat 77:616
Bueno-Ferrer C, Hablot E, Garrigós MC, Bocchini S, Averous L, Jiménez A (2012) Polym Degrad Stab 97:1964
Palanisamy A, Rao BS, Mehazabeen S (2011) J Polym Environ 19:698
Khanderay JC, Gite VV (2017) Green Mater 5:109
Rajput SD, Mahulikar PP, Gite VV (2014) Prog Org Coat 77:38
Ni B, Yang L, Wang C, Wang L, Finlow DE (2010) J Therm Anal Calorim 100:239
Bullermann J, Friebel S, Salthammer T, Spohnholz (2013) Prog Org Coat 76:609
Jiang L, Xu Q, Hu CP (2006) J Nanomater 2006:1
Liu X, Xu K, Liu H, Cai H, Su J, Fu Z, Guo Y, Chen M (2011) Prog Org Coat 72:612
Li Y, Noordover BAJ, van Benthem RATM, Koning CE (2014) Eur Polym J 52:12
Li Y, Noordover BAJ, van Benthem RATM, Koning CE (2015) Prog Org Coat 86:134
Fertier L, Koleilat H, Stemmelen M, Giani O, Joly-Duhamel C, Lapinte V, Robin JJ (2013) Prog Polym Sci 38:932
Mishraab V, Mohantya I, Patel MR, Patel KI (2015) Int J Polym Anal Charact 20:504
Chang CW, Lu KT (2013) Prog Org Coat 76:1024
Zhang P, Zhang X, Dai J (2011) Polym Mater Sci Eng 10:23
Llorente O, Fernández-Berridi MJ, González A, Irusta L (2016) Prog Org Coat 99:437
Jiang L, Xu Q, Hu CP (2006) J Nanomater 14906:1
Santiago A, Martin L, Iruin JJ, Fernández-Berridi MJ, González A, Irusta L (2014) Prog Org Coat 77:798
Acknowledgements
The authors acknowledge the University of the Basque Country UPV/EHU (UFI 11/56), the Diputación Foral de Gipuzkoa (OF218) and, the Basque Government (Ayuda a grupos de investigación del sistema universitario vasco IT618-13) for the funding received to develop this work. Technical and Human support provided by Macrobehavior-Mesostructure-Nanotechnology and NMR SGiker services of UPV/EHU is also gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Etxaniz, I., Llorente, O., Aizpurua, J. et al. Dispersion Characteristics and Curing Behaviour of Waterborne UV Crosslinkable Polyurethanes Based on Renewable Dimer Fatty Acid Polyesters. J Polym Environ 27, 189–197 (2019). https://doi.org/10.1007/s10924-018-1334-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10924-018-1334-0