
Abstract
Blending of lignin into thermoplastic materials presents a challenge due to the lack of dispersion and compatibility in the thermoplastic matrices. Kraft lignin was fractionated by methanol to homogenize its structure and molecular weight, and blended with poly(butylene adipate-co-terephthalate) (PBAT) and poly(lactic acid) (PLA). It was found through Fourier transform infrared spectroscopy that the lignin–polyester interaction involves aromatic group interactions as well as hydrogen bonding between the polymers. The differences in the intermolecular interactions led to high compatibility of lignin with PBAT and low compatibility with PLA as reflected by glass transition temperature shifts on the differential scanning calorimetry (DSC) curves. The DSC study also indicated that the methanol soluble lignin (MSL) fraction interacts with both PLA and PBAT, but no sign of interaction was evident between PLA and PBAT, which is reflected in the scanning electron microscope images depicting the morphology of the ternary blend. The resulting tensile properties showed retention of toughness at 30 % lignin content, and bridging of stress between PLA and PBAT by MSL.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
As an industrial by-product, lignin is produced at a rate of 70 million tons per annum, part of which is recycled back into the pulping process to provide energy for boiler recovery [1]. However pulping processes such as the Kraft process are bottlenecked by the accumulation of lignin not recycled back into the process and has little commercial value [2]. Attempt to find commercial applications for lignin as a thermoplastic material has been an ongoing study for the last few decades. The integration of lignin in the thermoplastic applications involves obtaining the best compatibility and dispersion of lignin in the matrix to optimize stress-transfer within the blend system. In theory, polymers which are polar in nature have the advantage of being able to create stronger intermolecular bonds with lignin compared to their non-polar counterparts [2]. Polyesters such as poly(lactic acid) (PLA), polyhydroxy alkanoates (PHAs), and poly(butylene succinate) (PBS) have carbonyl groups from their ester functionalities that can create hydrogen bonds with the hydroxyl groups of lignin. However, since the structure of lignin is highly heterogeneous in nature, weak interaction between some lignin compounds and the thermoplastic matrix would cause reduction in the overall performance [2]. Additionally, the heterogeneous structure would also cause inconsistencies in performance, which is undesirable for commercial applications [3]. Fractionation of lignin into more uniform compounds would negate this problem.
A number of lignin fractionation and solvent extraction studies have been conducted to further understand the heterogeneity of lignin. Both aqueous and a number of organic solvents have been used to fractionate lignin, along with ultrafiltration processes [4–18].
It has been shown that lignin compounds extracted by solvent with weak or moderate hydrogen bonding capability such as dichloromethane [4, 5], ethyl ether [9], and ethyl acetate [8] tend to be much lower in molecular weight. A gel permeation chromatography (GPC) study of these compounds showed that they have a low polydispersity (1.4–2.1), with a number average molecular weight (M n) of <800 g/mol for softwood Kraft lignin [18]. Based on the molecular weight, it has been concluded that these fractions are monomers and oligomers of the phenyl propane molecule that make up lignin [9]. Other commonly used solvents are propanol and iso-propanol [13, 14]. The use of alcohols such as methanol, ethanol, and propanol as a fractionating solvent extracts lignin molecules with an average M n of 440–3,300 g/mol; however they have a much higher polydispersity index (1.7–7.2) compared to dichloromethane, which also leads to higher yields of 33–53 %. The resulting material is more heterogeneous than the dichloromethane fraction [4, 5, 18]. Apart from differences in molecular weight, the fractions also have variations in chemical structure, geometric shape, and functional groups.
A number of blends of lignin and thermoplastic polyesters have been studied [19–25]. Blends of hardwood lignin with PLA [20], bagasse lignin with poly(hydroxyl butyrate) (PHB) [19], and lignin with PBS [22] each showed the presence of intermolecular interactions between each polymer and lignin. Blends of lignin with aliphatic–aromatic co-polyesters such as poly(butylene adipate-co-terephthalate) (PBAT) [25] and poly(ethylene terephthalate) (PET) [26, 27] have also shown good compatibility. The blends of Alcell, sisal, and abaca lignin with PBAT have shown retention of toughness and slight improvements in yield stress and tensile modulus, with lignin particle size ranging from 400 to 2,500 nm [25]. A melt spun blend of hardwood Kraft lignin and PET showed a single glass transition temperature (T g) which gradually shifts from the T g of PET to that of the lignin, in addition to shifts observed in the Fourier Transform Infrared (FTIR) spectra, indicating good compatibility [27].
The study on the blend of a synthetic polymer and fractionated lignin is much less common. Poly(vinyl chloride) (PVC) was blended with soda lignin which was successively fractionated with iso-propanol/ethanol mixture and methanol correlated the dispersion of lignin with its molecular weight [15]. The blend of PHB with bagasse soda lignin that was successively fractionated with diethyl ether and methanol found that the methanol soluble fraction increased the miscibility from the unfractionated lignin [19]. The blend of polypropylene (PP) with methanol and propanol insoluble fractions on the other hand reduced dispersion, which again shows the correlation between dispersion and molecular weight [3].
The aim of this paper is to study the effect of fractionated lignin in a thermoplastic blend which poses a challenge in the form of dispersion and compatibility. Hence, lignin fractions separated by methanol will be studied in a blend system with binary and ternary blends of PLA and PBAT matrices. The thermal and mechanical properties as well as morphological characterization of the resulting lignin will be studied.
Experimental
Materials
Injection grade poly (lactic acid) (PLA) Ingeo 3251D (specific gravity 1.24, melt flow rate 35 g/10 min at 190 °C and 2.16 kg loading, and a relative viscosity of 2.5) was purchased from Natureworks LLC, Minnetonka, Minnesota, USA. Poly(butylene adipate-co-terephthalate) (PBAT) with a grade name of Biocosafe 2003F (specific gravity 1.26, melt flow rate of ≤20 g/10 min) was purchased from Xinfu Pharmaceuticals, China. Softwood Kraft lignin Indulin AT was generously donated by MeadwestVaco, Richmond, Virginia, USA. The remaining solvents and reagents were purchased from Sigma-Aldrich Co.
Lignin Fractionation
Lignin fractionation was conducted by mixing lignin and methanol at a concentration of 0.6 g lignin/mL methanol at room temperature, filtered, and then remixed with the same quantity of methanol for a second time. The soluble fraction was collected and mixed with the same volume of HCl solution at pH 2.0, filtered, and rewashed with water to remove excess HCl. The insoluble fraction was washed with HCl solution and water. Both fractions were dried in a vacuum oven at 80 °C overnight to remove moisture and stored in a desiccator.
Blend Preparation
Prior to processing, PLA, PBAT, and lignin were dried in a convection oven at 80 °C for 6 h to remove moisture from the resin. Compounding and injection moulding of the blend was conducted using DSM Xplore 15 mL Micro-Compounder and 12 mL injection moulding machine. Compounding of the blend was done with co-rotating twin screw extruder with a processing temperature of 170 °C for all three processing zones, and a screw speed of 100 rpm for 3 min. Injection moulding was done with a holding temperature of 170 °C, mould temperature of 30 °C, injection pressure of 6 bars for 6 s, and holding and packing pressures of 6 bars for 6 s each. Conditioning of the samples was done for 48 h at temperature of 23 °C and 50 % relative humidity.
Characterization
Tensile and flexural information were obtained using Instron Universal Testing Machine Instrument Model 3382. The tensile was conducted according to ASTM D638 with at least 5 samples per test. A crosshead speed of 50 mm/min for tensile tests was used as recommended by the respective standards to achieve break less than 5 min.
Melt temperature (T m), crystallization temperature (T c) and glass transition temperature (T g), of the material were determined using the TA Instrument differential scanning calorimeter (DSC) Q200. The samples were prepared by placing 5–10 mg of sample in an aluminum pan. The DSC sample undergoes a heat/cool/heat cycle at a ramp rate of 10 °C/min from −50 °C to 170 °C, cooled back to −50 °C and finally heated to 250 °C under a nitrogen flow rate of 50 mL/min. Analysis of data obtained from the unit was done using TA Universal software.
A scanning electron microscope (SEM), HITACHI S-570, Japan, was utilized to examine the fracture surfaces of impact samples to observe the interaction between lignin and the polymer matrix. The tensile and impact samples were prepared by sputtering gold particles in order to increase electron conductivity on the surface of the sample. Furthermore, due to the sensitivity of PLA to heat, the electron beam was shot at an intensity of 10 kV to reduce the deformation on the sample surface.
To obtain a fracture sample without the effect of elongation, a cryo-fracture method has been adopted. The samples are notched and left inside liquid nitrogen for at least 30 min, followed by fracturing on the notched site. To observe the dispersion and particle size distribution within the blend, the fracture surface was exposed to an aqueous NaOH solution at pH 10 overnight to remove lignin from the SEM sample.
The Fourier transform infrared (FTIR) spectroscopy was conducted on a Thermo Scientific Nicolet 6700 FT-IR with a Smart Orbit attachment. Calibration is conducted with no sample loaded on the beam path, and the spectra recorded and averaged over 64 readings. Analysis of sample is conducted by loading a fine powder of the sample onto the platform, and the spectra is recorded and averaged over 64 readings.
Results and Discussions
Analysis of Lignin Fractions
The extraction process separates the Kraft lignin (KL) into two fractions: the methanol soluble lignin (MSL) and the methanol insoluble lignin (MIL). Upon extraction, the yield of the methanol soluble fraction was found to be consistently at 29.6–34.2 wt% of the overall initial lignin weight, a methanol insoluble fraction of 56.8–61.4 wt%, and a weight loss of approximately 9 wt%, similar to the yield found in literature [3, 18, 19].
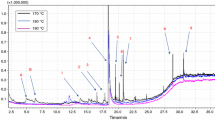
Since lignin behaves more similarly to a thermoset than a thermoplastic due to its high degree of crosslinking, a DSC analysis only presents a single glass transition temperature which is a function of molecular weight, chemical structure, degree of crosslinking, and intermolecular interactions [6]. As seen in Fig. 1, each lignin exhibits a single T g. The methanol insoluble fraction shows a T g of 186.7 °C, while the soluble fraction shows a T g of 120 °C. The difference in T g of the two fractions have been shown to be an effect of molecular weight differences [14, 18, 19]. The original lignin shows a T g intermediate of the two fractions. However, the T g of KL occurs at a relatively narrow range for a material consisting of two different fragments with a T g difference of more than 60 °C, indicating that the lignin fractions form intermolecular bonds typical of two miscible polymer system [19]. The fact that the fragments form intermolecular bonds will ultimately affect the ability of lignin to disperse and the method of interaction with the polymer matrix during melt processing [15].

Differential scanning calorimetry curves of Kraft lignin and its fractions
The FTIR analysis shown in Fig. 2 shows the differences in the chemical structures of KL, MSL, and MIL. The reference spectra were taken from Kubo et al. [28] starting from the higher wavenumber, the band occupying the wavenumber range of 3,100–3,600 cm−1 is produced by O–H stretching. The MSL band closely follows that of the original KL structure; however the MIL band is broader towards the higher wavenumber. The intensity of the peak at 2,700 to 3,000 cm−1 was attributed to C–H stretching in methyl and methylene groups. The band at ~1,704 and 1,205 cm−1 corresponds to the unconjugated stretching of the C=O and C–O bond, respectively. The intensity of the carbonyl peak is much higher for MSL compared to MIL which is likely responsible for the solubility of MSL in methanol due to the formation of hydrogen bonds between carbonyl and alcohol. The band at ~1,590 cm−1 which is caused by the aromatic skeletal vibration, is much narrower in the MSL fraction and much wider and intense in the MIL fraction. The second aromatic skeletal vibration peak at ~1,510 cm−1 is reduced in the MIL which also corresponds to the reduction of the band at ~1,265 cm−1, which is attributed to guaiacyl ring breathing with C–O stretching, which indicates a lower concentration of guaiacyl monomer in the MIL structure. Such heterogeneity in the chemical structure may have a large influence in the degree of interaction between the lignin molecule and the thermoplastic matrix.

FTIR analysis of Kraft lignin (KL), methanol insoluble lignin (MIL), and methanol soluble lignin (MSL)
Thermal Properties of Lignin Blends
The differential scanning calorimetry (DSC) curves of neat PBAT and PLA, binary blends of PBAT/PLA, PLA/MSL, PBAT/MSL, and the ternary blends of MSL can be seen in Fig. 3. PLA has a T g of 61.6 °C, a melting temperature approximately 170.0 °C, and undergoes crystallization during cooling and cold crystallization during heating at a temperature of 102.0 °C as observed in Fig. 3 [29]. On the other hand, PBAT shows a low T g of −34.0 °C, and a melting temperature of 117.5 °C, but no crystallization. The resulting blend of PLA and PBAT behaves as expected, with PBAT T g remaining relatively the same at −34.8 °C, PLA T g at 60.7 °C and a slight reduction in cold crystallization temperature of PLA due to the solid PBAT, as has been previously reported in literature [30].

DSC of different blends of PLA, PBAT, and lignin
The PLA/MSL lignin showed a slightly different behavior compared to neat PLA. Minor changes in T g can be seen from 61.6 to 59.5 °C, a behavior which persists in the ternary MSL blend which may indicate a plasticization effect of the low molecular weight lignin fraction towards PLA [29]. The decrease in the T g was also due to the formation of hydrogen bonding between the hydroxyl phenolic groups of lignin with the carbonyl groups of PLA. These results agree with previous studies of PLA-lignin blends [20]. The increase in cold crystallization temperature from 102 to 109 °C and the reduced melting temperatures from 170.0 to 167.9 °C indicates that MSL is anti-nucleating PLA, retarding the crystallization of PLA [29]. Additionally, the addition of MSL caused PLA melting endotherm to start at a lower temperature, whereas neat PLA and PLA/PBAT blends showed a recrystallization exotherm prior to melting [31]. This behavior is not observed with the MIL fraction, which may indicate that the lower softening temperature of MSL causes mobility of MSL between PLA chains.
PBAT/MSL binary blend also showed a different behavior compared to both neat PBAT and PLA/PBAT blend. The T g of PBAT was increased from −33.9 to −11.4 °C which indicates miscibility between the PBAT and MSL phase. In addition to the increased T g, a reduction of PBAT melting temperature of 5 °C was also observed [19]. The T g of MSL is not observed here which may be because PBAT/MSL is exhibiting a single glass transition due to complete miscibility, or similar to the other blends, the change in heat flow from the glass transition of MSL is too subtle to be observed [19].
It can further be observed that the resulting behavior of the MSL binary blends is translatable to the ternary blends, i.e. the MSL ternary blend showed an increase in PBAT T g that is similar to the PBAT/MSL blend and PLA T g, T c, and T m that are similar to the PLA/MSL blend. This result may indicate that MSL is in contact with both PBAT and PLA, while PBAT and PLA are not in contact, which is confirmed by the morphological studies. The PBAT phase showed further increase in T g from −34 to −1 °C which may be caused by the different weight ratio of PBAT to lignin in the ternary blend (62/38) compared to the PBAT/MSL binary blend (70/30). The melting temperature of PBAT cannot be determined due to the fact that it occurs almost simultaneously with PLA cold crystallization, and is overwhelmed by the exotherm released during the cold crystallization due to the low crystallinity of PBAT [30].
Thermal properties of the ternary blends of the remaining two lignin showed different behavior compared to the MSL. The shift in T g of the PBAT phase in the KL (−26.0 °C) and MIL (−30.9 °C) blends are less prominent compared to the MSL, suggesting that there is little compatibility between MIL and PBAT, and that the shift in the KL blend is attributed to the MSL component [19]. Another prominent difference in the lignin fractions can be seen with the crystallization characteristics. As observed the KL and MIL fractions reduced the cold crystallization temperature of PLA from 102 to 95.1 °C for KL blend and 96.8 °C for MIL blend, which suggests the effect of nucleation [29]. As seen in Fig. 1, the MIL fraction has a T g of 187 °C, which means that it is still in solid state at the temperature range, acting as a nucleation site for PLA crystals.
FTIR Analysis of Lignin Blends
The FTIR analysis of the blends can be seen in Fig. 4. The hydroxyl peak of MIL and MSL which are originally found at 3,374 and 3,392 cm−1 respectively, have been found to shift to 3,414 cm−1 for PBAT/MIL, 3,426 cm−1 for PBAT/MSL, and 3,504 cm−1 for PLA/MSL, indicating the intermolecular interaction associated with the hydroxyl groups of lignin [27]. PLA and PBAT have different carbonyl bands due to differences in neighboring chemical structures. PLA has a carbonyl stretching at 1,747 cm−1 while PBAT has a carbonyl stretching at 1,712 cm−1. The carbonyl stretching of both polymers have not been affected much after the addition of both MIL and MSL. In the lower wavenumbers, shifts in the aromatic skeletal vibration peaks can also be seen. The first aromatic skeletal vibration which is 1,591 cm−1 for MIL and 1,593 cm−1 for MSL have been found to shift to 1,596 cm−1 for PBAT/MIL, 1,597 cm−1 for PBAT/MSL, and 1,596 cm−1 for PLA/MSL. The second aromatic skeletal vibration is located at 1,508 cm−1 for MIL and 1,511 cm−1 for MSL. These peaks have also been shifted to a higher wavenumber of 1,512 cm−1 for PBAT/MIL, 1,514 cm−1 for PBAT/MSL, and 1,515 cm−1 for PLA/MSL.

FTIR of lignin, PLA, PBAT, and their binary blends
These shifts in spectra indicate the interactions between lignin and the polyesters are more than just hydrogen bonding between hydroxyl groups of lignin and carbonyl groups of the polyesters. A review by Barlow et al. [32] have shown that the aromatic groups of polymers can often interact with other functional groups to lead to miscibility of different polymers. On top of hydrogen bonding, the combination of lignin and PLA can form other intermolecular interactions such as π-hydrogen bonding between the aromatic structure of lignin and the methyl of PLA, n–π complex between aromatic of lignin and the carbonyl of PLA, and dipole–dipole interactions [32]. The combination of lignin (both MSL and MIL) and PBAT can form the same intermolecular bonds as lignin/PLA; however, the aromatic ring of PBAT can offer additional interactions such as π hydrogen bonding between the hydroxyl group of lignin and the aromatic ring of PBAT, and π–π complex between the lignin aromatic structure and the PBAT aromatic structure. The π–π complex may also explain the high compatibility between lignin and other aromatic containing polymers such as PET as observed by Kubo et al. [27] and polystyrene as observed by Pouteau et al. [24] since it has been studied to be a relatively strong and stable intermolecular interaction [33]. Additionally, the presence of carbonyl groups on MSL adds the possibility of n–π complex between carbonyl of lignin and aromatic of PBAT [32]. As such, it can be seen why the presence of aromatic structures in both lignin and PBAT may be an important factor in determining the compatibility between lignin and the matrix.
Blend Morphology
The SEM images of the PLA/PBAT/lignin blends can be seen in Fig. 5. Figure 5a represents the cryo-fractured surface of neat PLA/PBAT blend at a 30/70 weight ratio. The immiscibility of the two phases can be observed, with PBAT as the continuous phase and PLA as the discontinuous phase. PLA which is in the form of white beads with a particle size of approximately 1 µm shows almost no wetting with the PBAT phase indicating very low compatibility. Figure 5b shows the same PLA/PBAT blend with 1 wt% MSL added during processing. As observed, although the particle size of the PLA beads remained unchanged, the addition of MSL reduces the immiscibility of the two phases in that wetting of the PLA beads can be seen [34, 35]. Figure 5c shows the schematic of the blend where the PLA beads are represented by the blue spheres with relatively even particle size, and PBAT is represented by the yellow background.

SEM images of PLA/PBAT (30/70) binary blend and PLA/PBAT/lignin ternary blends. a PLA/PBAT, b PLA/PBAT with 1 % MSL, c schematic of PLA/PBAT, d PLA/PBAT/KL, e solvent extracted PLA/PBAT/KL, f schematic of PLA/PBAT/KL, g PLA/PBAT/MSL, h solvent extracted PLA/PBAT/MSL, i schematic of PLA/PBAT/MSL, j PLA/PBAT/MIL, k solvent extracted PLA/PBAT/MIL, l schematic of PLA/PBAT/MIL
The SEM images of the PLA/PBAT/KL blends are shown in Fig. 5d–f. Figure 5d is of the cryo-fractured surface of the PLA/PBAT/KL with a weight ratio of 21/49/30. The fracture surface showed a very rough terrain. A comparison of Fig. 5a, d shows that there is no clear phase separation between the components in D, while A showed a distinct phase separation between PLA and PBAT. When lignin is extracted from the blend by selective solvent extraction using NaOH solution, the differences between the three components are much clearer, as shown in Fig. 5e. The spaces that were occupied by the lignin particles show a large lignin particle size distribution with lignin particles as large as 50 µm (not shown in the image) and lower than 1 µm. Such particles are likely lignin agglomerates that are formed by the formation of intermolecular bonds within lignin molecules. The large lignin molecules offer two obvious disadvantages towards the blend system. Firstly, the formation of large particles would mean that there is reduced surface area for stress-transfer between the matrix and lignin. Secondly, the lignin–lignin interaction is a brittle one, which leads to reduced toughness of the resulting blend. The space left behind by lignin also show evidence of wetting between lignin and PBAT as observed by the veins in the craters.
Figure 5e also shows gaps between PLA and PBAT phase that have been left by lignin, showing the placement of lignin between the two interfaces. This morphology can be explained by Fig. 5f where we have large particle size distribution of lignin depicted by the brown spheres, but the smaller lignin particles can be found in the interface between PLA and PBAT.
Additionally, the PLA phase comprised of larger spheres with sizes of up to 5 µm rather than small beads of 1 µm. This change in particle size is a shift in morphology towards the phase inversion of PLA and PBAT upon the addition of lignin. Utracki et al. [36] have shown that the morphology of two immiscible polymers is co-continuous when the volumetric ratio is equal to the viscosity ratio. Since the weight ratio of PLA and PBAT remained the same and lignin is only found in the PBAT phase, the shift towards the phase inversion would mean that the viscosity of PBAT has been reduced by the addition of lignin.
Comparing the KL ternary blend morphology in Fig. 5d with the MSL ternary blend morphology in Fig. 5g shows there is a difference in the roughness of the terrain. Upon extraction (Fig. 5h), we could see that the average particle size of lignin was much smaller compared to the KL blend, with particle size <1 µm. Similar to the KL blend, the extraction of lignin from the blend reveals the structure of PLA and PBAT phase. However, since there is a higher concentration of the MSL fraction, the reduction of viscosity of the PBAT phase is even higher, resulting in a further shift towards co-continuous morphology, creating elongated PLA morphology. The extraction of lignin also proved the existence of lignin between the PLA/PBAT interfaces, which is further reinforced by the DSC data.
Similar to the KL blend, the MIL blend also showed an extremely rough terrain as seen in Fig. 5j. The extracted surface morphology (Fig. 5k) however, showed the difference between the KL and MIL blends. Unlike the KL blend, the MIL blend does not have the smaller lignin particles associated with the MSL blend. Additionally, the craters left by the extracted lignin showed no signs of wetting between PBAT and MIL which confirms the lack of interaction between PBAT and MIL found in the DSC study. The PLA phase can be found as spheres inside the lignin craters, which shows that despite the reduced interaction between MIL and PBAT, the solubility of PLA is still closer to that of MIL than PBAT.
Mechanical Properties
The stress–strain curves for the blends are shown in Fig. 6 and the mechanical properties values are displayed in Table 1. PBAT is an extremely tough polymer, with an elongation of over 500 %. The tensile strength of PBAT stems from the fact that at higher elongations, the PBAT chains reorganize to undergo stress induced crystallization, thereby increasing the force required to break [37] as observed in the stress–strain curves in Fig. 6. Although PBAT has high toughness, its yield strength and modulus is still lacking.

PLA/PBAT/lignin blends stress–strain curves
The PBAT/KL blend fares much better with regards to yield strength and modulus, due to the addition of solid filler which prevents polymer chains from slipping, thereby reducing movement and making the blend stiffer [38]. Additionally, since PBAT is still a major phase, it still influences the elongation of PBAT/lignin blends. As seen in the stress–strain curve of the PBAT/KL blend, elongation is severely reduced compared to neat PBAT. Since lignin restricts the movement of the polymer chain, chain entanglement and the degree of stress-induced crystallization of the PBAT matrix is reduced, reducing elongation and tensile strength [21]. Lignin agglomerates would also play a role in reducing the tensile properties of the blend since the lignin–lignin interaction is very brittle, and the large particle size would reduce the surface area for stress transfer between lignin and the PBAT matrix. The yield strength was improved by the addition of KL as seen in the stress–strain curves. Previous studies by Nitz et al. [25] on blend of PBAT with Alcell, sisal, and abaca lignin showed the same increase in yield stress, modulus, and retention of toughness up to 50 % of lignin by weight.
The PBAT/MSL blends on the other hand shows a slightly different behavior. The retention of elongation indicates that lignin is well dispersed within the PBAT matrix, since lignin is brittle in nature. The highly dispersed lignin particle and good interaction between lignin and PBAT as observed in the DSC study, means that there is good energy transfer between the two phases. However, the tensile strength at break is lower compared to neat PBAT as seen in Table 1, which is again due to the inability of the PBAT/MSL blend to undergo stress induced crystallization.
The addition of PLA to PBAT improves the yield strength of the blend, whereas the stress at break and the effect of stress induced crystallization remained relatively similar. Since PBAT is still the continuous phase in the 30/70 PLA/PBAT blend, its properties is still highly influenced by the properties of PBAT. With constant strain, the material first undergoes yield, followed by necking, then elongation and stress induced crystallization of the PBAT phase. The behavior of the PLA/PBAT is consistent with reported results where phase separation and incompatibility of the PLA/PBAT phases caused reduction in mechanical properties from predicted theoretical values [35].
The KL and MIL ternary blends with PLA and PBAT showed very similar tensile behavior. This similarity between the KL and MIL blends with the MSL blend, indicate that the mechanical property of the KL blend is highly influenced by the MIL component of the kraft lignin. It can also be noted that in the KL and MIL ternary blends, necking was not observed after yield, which is likely attributed to the brittle PLA-MIL interaction and low dispersion of MIL in PBAT. The yield in the stress–strain curve of the KL and MSL ternary blends are not as abrupt as the PLA/PBAT binary blend, which may suggest that MSL is bridging the stress transfer between the two incompatible phases as observed in the morphological study of the ternary blends, which was not observed with the MIL blend.
The MSL ternary blend exhibits elongation similar to that of the original PLA/PBAT blend. Note that the yield strength for all ternary lignin blends is approximately 2/3 of its original value as observed in Table 1, which can be directly correlated to the reduction in overall PLA content from 30 to 21 wt%. This lack of change suggests that the lignin fractions have little effect on the properties of the PLA phase, which is further supported by the DSC thermograms showing very little change in the thermal properties of the PLA phase. Similar to the PLA/PBAT blend, the MSL blend showed stress induced crystallization characteristic of the continuous PBAT phase.
The lack of change with the tensile properties associated with the PLA phase suggests that in the ternary blends, lignin does not have much effect on the PLA domains. On the other hand, the changes observed with the PBAT/lignin binary blends suggest interaction between PBAT and lignin. However, the significant reduction in impact strengths and the smooth transition between the yield of PLA and drawing of PBAT on the ternary blends show that there is an interface where PLA and lignin interacts as observed in the SEM images and DSC study. The lignin particulates are well dispersed within the PBAT matrix, but see very little dispersion in the PLA domain, reflecting the differences in solubility of lignin in PLA and PBAT. However, some hydrogen bonding does occur with the hydroxyl group of lignin and the carbonyl groups of PLA [39], which forms an interface that bridges the PLA and PBAT domains.
Conclusions
The blends of lignin and its methanol fractionated components showed the effect of heterogeneity on the ability of lignin to be blended into a thermoplastic matrix. Fractionation of Kraft lignin in methanol showed that the soluble and insoluble lignin fractions have pretty extensive differences thermally, chemically, and physically. These differences between the fractions also lead to stark contrast between their dispersion and compatibility in the bioplastic matrices. The PBAT/lignin binary blends showed large variation in dispersion and compatibility of the two lignin fractions with PBAT. DSC studies showed significant changes in the glass transition temperature of the PBAT phase, pointing to compatibility of PBAT and MSL, which is reflected in the mechanical properties showing retention of elongation and toughness, with improvements in yield strength. Additionally, FTIR studies showed the presence of hydrogen bonding as well as aromatic interactions which may explain the differences in the intermolecular interactions between PBAT/lignin and PLA/lignin.
The addition of PLA to the PBAT/lignin blend showed improvements in strength and modulus as expected. Additionally, compared to the neat PLA/PBAT blend, the ternary blends showed more continuous behavior under tensile test, indicating lignin is bridging the two incompatible PLA/PBAT phases, which was confirmed by the SEM images and DSC studies. The KL and MIL blends showed similar elongation values indicating that there is some interaction between the polyesters and lignin as reflected by the lack of compatibility observed in the FTIR and DSC study. All of these results indicated that the fractionation of lignin by dissolution in methanol into more homogeneous fractions offers better dispersion and more consistent properties to PLA-PBAT blends.
References
Satheesh Kumar MN, Mohanty AK, Erickson L, Misra M (2009) J Biobased Mater Bioenergy 3:1
Doherty WOS, Mousavioun P, Fellows CM (2011) Ind Crops Prod 33:259
Pouteau C, Dole P, Cathala B, Averous L, Boquillon N (2003) Polym Degrad Stab 81:9
Yoshida H, Morck R, Kringstad Knut P, Hatakeyama H (1987) Holzforschung—Int J Biol Chem Phys Technol Wood 41:171
Morck R, Reimann A, Kringstad Knut P (1988) Holzforschung—Int J Biol Chem Phys Technol Wood 42:111
Glasser WG, Sarkanen S (1989) In: Symposium at the 195th national meeting of the American chemical society, Toronto, Ontario, Canada, June 5–11, American Chemical Society, pp 545
de Oliveira W, Glasser WG (1994) Macromolecules 27:5
Thring RW, Griffin SL (1995) Can J Chem 73:629
Thring RW, Vanderlaan MN, Griffin SL (1999) J Wood Chem Technol 16:139
Ghosh I, Jain Rajesh K, Glasser Wolfgang G (1999) Lignin: historical, biological, and materials perspectives. American Chemical Society, Washington, DC, pp 331
Sun R, Tomkinson J, Griffiths S (2000) Int J Polym Anal Charact 5:531
Pucciariello R, Villani V, Bonini C, D’Auria M, Vetere T (2004) Polymer 45:4159
Leger CA, Chan FD, Schneider MH (2010) Bioresources 5:2239
Yue X, Chen F, Zhou X (2012) J Macromol Sci Phys 51:242
Yue X, Chen F, Zhou X, He G (2012) Int J Polym Mater 61:214
Tunc MS, Chheda J, van der Heide E, Morris J, van Heiningen A (2014) Holzforschung 68:401
Cui CZ, Sun RK, Argyropoulos DS (2014) ACS Sustain Chem Eng 2:959
Morck R, Yoshida H, Kringstad KP, Hatakeyama H (1986) Holzforschung 40:51
Mousavioun P, Doherty WOS, George G (2010) Ind Crops Prod 32:656
Li J, He Y, Inoue Y (2003) Polym Int 52:949
Li J, He Y, Inoue Y (2001) Polym J 33:336
Sahoo S, Misra M, Mohanty AK (2011) Compos A 42:1710
Kubo S, Kadla JF (2005) J Appl Polym Sci 98:1437
Pouteau C, Baumberger S, Cathala B, Dole P (2004) C R Biol 327:935
Nitz H, Semke H, Mulhaupt R (2001) Macromol Mater Eng 286:737
Kubo S, Kadla JF (2005) J Polym Environ 13:97
Kadla JF, Kubo S (2004) Compos A 35:395
Kubo S, Kadla JF (2005) Biomacromolecules 6:2815
Auras R, Loong-Tak L, Selke SEM, Tsuji H (2010) Poly(lactic acid). Synthesis, structures, properties, processing, and applications. Wiley, NY
Jiang L, Wolcott MP, Zhang JW (2006) Biomacromolecules 7:199
Yeh J-T, Tsou C-H, Huang C-Y, Chen K-N, Wu C-S, Chai WL (2010) J Appl Polym Sci 116:680
Barlow JW, Paul DR (1981) Annu Rev Mater Sci 11:299
Hunter CA, Sanders JKM (1990) J Am Chem Soc 112:5525
Lee S, Lee Y, Lee JW (2007) Macromol Res 15:44
Zhang N, Wang Q, Ren J, Wang L (2009) J Mater Sci 44:250
Utracki LA (1991) J Rheol 35:1615
Chivrac F, Kadlecova Z, Pollet E, Averous L (2006) J Polym Environ 14:393
Nyambo C, Mohanty AK, Misra M (2010) Biomacromolecules 11:1654
Ouyang W, Huang Y, Luo H, Wang D (2012) J Polym Environ 20:1
Acknowledgments
The financial support from the Natural Sciences and Engineering Research Council (NSERC), Canada Lignoworks Network to carry out this research is gratefully acknowledged. Authors also acknowledge MeadWestvaco for providing the lignin samples for this research.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Mohamed A. Abdelwahab was on leave from Department of Chemistry, Tanta University, Tanta, 31527, Egypt.
Rights and permissions
About this article

Cite this article
Chen, R., Abdelwahab, M.A., Misra, M. et al. Biobased Ternary Blends of Lignin, Poly(Lactic Acid), and Poly(Butylene Adipate-co-Terephthalate): The Effect of Lignin Heterogeneity on Blend Morphology and Compatibility. J Polym Environ 22, 439–448 (2014). https://doi.org/10.1007/s10924-014-0704-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10924-014-0704-5