
Abstract
Hypercrosslinked porous polymers have become a hotspot of research by virtue of their high specific surface area, mild synthesis conditions, and wide source of monomers, which are promising adsorbents for water treatment. In this paper, 4-vinylbenzylchloride (VBC), acrylic acid (AA) and divinylbenzene (DVB) were used to prepare crosslinked microspheres as a template by reflux precipitation polymerization. On this basis, porous microspheres were prepared by hypercrosslinking reaction using FeCl3 as catalyst. Next, excess FeCl3 was used as iron sources for the next solvothermal reaction to prepare magnetic hypercrosslinked P(VBC-DVB-AA)@Fe3O4 microspheres (MHCPMs). The elution step of the hypercrosslinked product was omitted, while magnetic Fe3O4 was introduced. MHCPMs have uniform particle size, high specific surface area, superparamagnetism and abundant functional groups. After synergistic preparation, the specific surface area of the MHCPMs-60 was still as high as 654.56 m2/g, and the saturation magnetization strength reached 29.0 eum/g. The magnetic microspheres could be directly aggregated and separated in the presence of an applied magnetic field. In addition, MHCPMs were well dispersed in water after vigorous shaking and could be used to adsorb the cationic dye quickly and efficiently. The adsorption results for methylene blue (MB) well conformed to the Langmuir model (R2 = 0.999) with a maximum adsorption of 214.26 mg/g and followed the pseudo-first-order kinetic model. In addition, the MHCPMs-60 showed good reusability and stability over seven cycles. Therefore, this study proposed a synergistic strategy for the preparation of magnetic porous polymer microspheres, which exhibited a significant potential application value in many fields.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
In recent years, industrial and domestic wastewater generated by rapid industrialization and rapid population growth have caused serious water pollution [1]. Pollutants include antibiotics [2], oil spills [3], organic pesticides [4,5,6], dyes [7,8,9], and others [10,11,12]. Dyes are important materials in all textile industries and are mainly used to add color to fabrics. It has been reported that about 40,000 tons of industrial dye wastewater is transferred from different enterprises every year [13]. These pollutants can have significant negative impacts on the ecosystem and human health [14]. Therefore, it is necessary to treat the wastewater before discharge.
In order to effectively treat dye wastewater, a number of separation technologies have been developed, including physical methods (adsorption, membrane separation, magnetic separation), chemical methods (electrochemistry, photochemical and photocatalytic oxidation, ozone oxidation) and biological methods (anaerobic, aerobic, anaerobic–aerobic combination) [15]. Among them, adsorption method is widely used due to its low cost, convenience and ease of operation [16]. Similarly, many materials have been used as adsorbents, such as porous carbon [17], chitosan [18, 19], zeolites [20], and ion exchange resins [21]. During the adsorption process, different compositions and structures of adsorbents produce different interactions with dyes, such as van der Waals forces, electrostatic interactions and π-π interactions, etc., which affect their adsorption effects [22, 23]. In general, the porous structure and active site of the adsorbent are considered to be the key influencing factors on the adsorption behavior [24]. For example, Hao et al. [17] prepared multistage porous carbon using cellulose as a carbon precursor with a heat treatment process. And their specific surface area and pore size were regulated by changing the holding temperature to obtain higher adsorption capacity. Porous inorganic materials possess high specific surface area and controllable pore structure. However, they lack active sites and need to be functionalized or other means to improve their performance [25].
Recently, porous organic polymers (POPs) have received an increased level of research interest due to their special properties achieved by combining the advanced properties of both porous materials and polymers [26]. POPs types such as hypercrosslinked polymers (HCPs) [27], polymers of intrinsic microporosity (PIMs) [28], conjugated microporous polymers (CMPs) [29] and covalent organic frameworks (COFs) [30] have been developed and applied to gas storage and separation [31], as supports for catalysis [32], adsorbents [33], energy storage [34], sensors [35], etc. However, the synthesis of CMPs and some other POPs requires the use of complex monomer units, expensive transition metal catalysts, or harsh reaction conditions [36], resulting in high production costs. Among various POPs, HCPs can be prepared using cheap FeCl3 catalyzed Friedel–Crafts alkylation reaction. But after synthesis, HCPs have to be washed several times to remove the catalyst in their structure. For example, Liu et al. [37] first prepared a lignin-based copolymer (LCP) precursor by graft copolymerization of organosolv lignin with 4-vinylbenzylchloride (VBC) and divinylbenzene (DVB) monomers. Then, the in situ Friedel–Crafts reaction was implemented in the structure of the precursor copolymer to obtain the crude product. The crude product was then filtered, washed alternately with 1% HCl (v/v), ethanol and distilled water. Finally, they were purified in ethanol by a Soxhlet extractor to obtain the final product. In addition, HCPs can be modified by diverse functional monomers to provide more active sites while ensuring the porous structure of the adsorbents [38]. When applying HCPs to the treatment of dye wastewater, some hydrophilic groups can be introduced on the surface of the hydrophobic adsorbent to improve its adsorption behavior [39]. Liu et al. [40] reported that the HCPs (MPD-HCP) with the introduction of building units containing amino functional groups, which were able to efficiently extract nitroimidazole from water, beverage, and honey samples due to their hydrophilic, microporous structure, and active site.
However, collecting the adsorbent from the dispersed medium is a difficult task, which may bring about secondary contamination and cause many inconveniences in practical applications. Designing magnetic adsorbents may be an effective and easy way to rapidly collect adsorbents for further processing [41, 42]. Fe3O4 is one of the most widely used magnetic materials. The common preparation methods of Fe3O4 nanoparticles include the co-deposition method [43], the mechanical balls grinding method, high-temperature thermal decomposition method [44], sol–gel method [45] and the solvothermal method [46]. The solvothermal method is a synthesis method of reaction precursor formed under certain reaction temperature and system pressure in vacuum or inert gas system with organic liquid as solvent. The solvothermal synthesis of Fe3O4 nanoclusters has the advantages of uniform size, strong controllability, good dispersion, high magnetization and low requirements for equipment performance, which have attracted the attention of many researchers. Therefore, by combining HCPs with magnetic Fe3O4, the magnetic porous polymer adsorbents not only have good adsorption properties, but also is easy to be recycled.
In this work, a synergistic and facile method was proposed to obtain carboxyl-functionalized magnetic hypercrosslinked porous P(VBC-DVB-AA)@Fe3O4 microspheres (MHCPMs). First, microspheres with different crosslinking degrees were prepared by reflux precipitation polymerization using 4-vinylbenzylchloride (VBC) and acrylic acid (AA) as monomers and divinylbenzene (DVB) as crosslinking agents. And the addition of AA provided abundant carboxyl functional groups for seed microspheres. On this basis, porous microspheres were prepared by Friedel–Crafts alkylation reaction with FeCl3 as catalyst. Then, the washing process of the hypercrosslinked products was omitted and the excess catalyst was used as an iron source for the next step of the solvothermal reaction to prepare MHCPMs. FeCl3 was partially reduced, resulting in the in-situ generation of Fe3O4 magnetic nanoclusters on the microspheres. Under the action of an external magnetic field, the magnetic microspheres can be directly aggregated and separated. And the effects of crosslinking degree of seed microspheres and catalyst dosage on the formation of the MHCPMs were investigated. Finally, the adsorption capacity of MHCPMs to methylene blue (MB) in wastewater was studied under different adsorption control parameters. The adsorption process was analyzed by isothermal and kinetic analysis, and its reusability was evaluated [47,48,49]. The experimental results indicated that the MHCPMs-60 could be used as an efficient, durable and sustainable adsorbent for water treatment [50, 51].
2 Experimental Section
2.1 Materials
4-vinylbenzylchloride (VBC), acrylic acid (AA), divinylbenzene (DVB), acetonitrile (ACN), toluene (TL), and methylene blue (MB) were purchased from Shanghai Macklin Biochemical Technology Co., Ltd. Polyvinylpyrrolidone (PVP) was commercially available from Yuanye Biotechnology Co., Ltd. (Shanghai, China). Azobisisobutyronitrile (AIBN) was obtained from Shanghai Titan Technology Co., Ltd. sodium acetate (NaAc), ethylene glycol (EG) and 1,2-dichloroethane (DCE) were provided by Shanghai Lingfeng Chemical Reagent Co., Ltd. Anhydrous ferric chloride (FeCl3), ferric chloride hexahydrate (FeCl3·6H2O), and sodium hydroxide (NaOH) were all supplied by Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China).
3 Preparation of P(VBC-DVB-AA) Microspheres (PMs)
A simple reflux precipitation polymerization method was used to synthesize P(VBC-DVB-AA) microspheres (PMs). First, 120 mL of ACN and 30 mL of toluene were added into a three-necked flask. Second, 1.5 mL VBC, 1.5 mL of AA and 4.5 mL of DVB were added to the three-necked flask with stirring. When the temperature of the reaction solution stabilized at 89 °C, 0.07 g of AIBN was added under the protection of nitrogen, and the polymerization reaction was carried out for 4 h [52]. Finally, the solution was centrifuged, washed three times with ethanol, and then dried in a vacuum drying oven to obtain P(VBC-DVB-AA) microspheres. According to the addition amount of monomer DVB (30%, 40%, 50%, 60% by volume), they were named as PMs-30,40,50,60, respectively. And the specific feeding quantity of monomers was tabulated in Table 1.
4 Preparation of Hypercrosslinked P(VBC-DVB-AA) Microspheres (HCPMs)
Polymerization of hypercrosslinked P(VBC-DVB-AA) microspheres (HCPMs) was performed as follows: First, 0.5 g of dried PMs were swelled in 20 mL of DCE for 12 h under magnetic stirring at 35 °C. 1.5 g of FeCl3 was ultrasonically dispersed in 20 mL of DCE and then added to the swollen mixture. Second, the original network was formed by stirring the mixture at 45 °C for 5 h via a Friedel–Crafts-type hypercrosslinking reaction. Next, the mixture was heated at 80 °C for 20 h to obtain the porous polymer microspheres [52, 53]. Finally, the obtained products were recovered without washing and dried under vacuum. Based on the previous PMs-30,40,50,60, the achieved hypercrosslinked microspheres were named as HCPMs-30, 40, 50, 60, respectively. And the specific feeding quantity was tabulated in Table 2.
5 Preparation of Magnetic Hypercrosslinked P(VBC-DVB-AA)@Fe3O4 Microspheres (MHCPMs)
Polymerization of magnetic hypercrosslinked P(VBC-DVB-AA)@Fe3O4 microspheres (MHCPMs) was performed as follows: Firstly, 0.5 g of the above unwashed HCPMs were dispersed in 80 mL EG by ultrasonication, and then 10 g of NaAc and 0.8 g of H2O were added. The mixture was transferred to a high-pressure reactor to heat up and react for a period of time. After cooling, the suspension was repeatedly centrifuged and washed with deionized water several times to obtain magnetic porous microspheres, which were named as MHCPMs-30 and MHCPMs-60 based on the difference of the template microspheres (the PMs-30 and PMs-60).
As a control experiment, magnetic P(VBC-DVB-AA)@Fe3O4 microspheres (MPMs) were achieved based on PMs-60 under the same reaction conditions. And the specific feeding quantity was tabulated in Table 3.
6 Adsorption Study of Dye
The adsorption capability of MHCPMs towards water soluble pollutants was evaluated based on the removal of MB from aqueous solutions. MHCPMs adsorbent (4 mg) were added to MB aqueous solutions with different initial concentrations (40 mL, 5–50 mg/L), and then each sample was shaken at specific temperature and pH for 6 h to reach adsorption equilibrium. The pH was adjusted using 0.1 M of both HCl and NaOH in a range of 2 to 12. The experiments were performed from 25℃ to 45℃. The adsorption time optimization was performed using 4 mL of 20 mg/L MB aqueous solution and 4 mg of MHPMs shaken with 200 rpm at 25℃. MHCPMs were removed magnetically and the remaining concentration of MB was analyzed by a UV-2600 spectroscope (Shimadzu, Japan) at a wavelength of 664 nm (maximum absorbance of MB). The dye removal efficiency (R) and the adsorption capacity (Qe) were calculated according to Eqs. (1) and (2)
where V (L) was the volume of the dye solution; C0 and Ce (mg/L) were mass concentrations of dyes before and after adsorption; m (g) was the mass of MHCMPs; and Qe (mg/g) was the adsorption capacity of MHCMPs.
To test the recycling performance of MHCPMs, continuous adsorption–desorption experiments were performed on MB solution. After the adsorption process, the adsorbent was separated using a magnet and then thoroughly washed with deionized water and ethanol. After drying at 60℃ overnight, the regenerative adsorbent was used again.
7 Characterization
Fourier transform infrared spectroscopy (FTIR) and UV–vis absorption spectroscopy were performed on a Nicolet-5700 spectroscope (Asheville) and a UV-2600 spectroscope (Shimadzu, Japan). Transmission electron microscopy (TEM, FEI, USA) at an acceleration voltage of 200 kV and field-emission scanning electron microscope (SEM, FEI Inspect F50, USA) operated at 12.5 kV were used to characterize the morphologies of materials. In order to obtain the elemental distribution map, energy-dispersive X-ray spectroscopy (EDX) was carried out in conjunction with TEM. The crystalline patterns of the magnetic microspheres were obtained using an Ultima IV powder X-ray diffraction (XRD, Shimadzu, Japan). Thermogravimetric analysis (TGA) was performed on a TG-209 F-3 instrument (NETZSCH, Germany) and the samples were heated to 800 °C in nitrogen gas at a rate of 20 °C/minute. The magnetic properties of the microspheres were measured by the VSM-7407 vibrating sample magnetometer (VSM, Lake Shore). The N2 adsorption isotherm (77 K), surface areas and pore size distribution of the porous microspheres were determined using an Quantachrome Autosorb IQ MP specific surface and porosity analyzer (BET, Micromeritics). The zeta potential of the samples was tested by a NanoBrook multi-angle particle size potential analyzer from Brookhaven, USA.
8 Results and Discussion
8.1 Characterization of Hypercrosslinked P(VBC-DVB-AA) Microspheres and Magnetic Hypercrosslinked P(VBC-DVB-AA)@Fe3O4 Microspheres
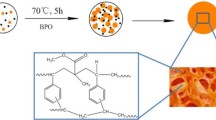
The steps required for the preparation of MHCPMs and MPMs are outlined in Scheme 1. First, P(VBC-DVB-AA) microspheres with different DVB contents were prepared by reflux precipitation polymerization. Next, the crosslinked microspheres were achieved by Friedel–Crafts alkylation reaction using FeCl3 as a catalyst, followed by centrifugation to remove the solvent and drying without washing. Then the hypercrosslinked microspheres were used as templates and the catalyst was used as iron sources. MHCPMs were obtained by in situ generation of Fe3O4 nanoparticles on the surface of the hypercrosslinked microspheres by solvothermal reaction. And as a control experiment, MPMs were obtained directly by solvothermal reaction of the crosslinked microspheres without hypercrosslinking process. In this synthesis of MHCPMs, FeCl3 was used both as a catalyst for the hypercrosslinking process and as sources of iron for secondary use, which simplified the experimental steps and saved resources.

Schematic Diagram of P(VBC-DVB-AA)@Fe3O4 Microspheres (MPMs) and Hypercrosslinked P(VBC-DVB-AA)@Fe3O4 Microspheres (MHCPMs)
In order to track the above synthesis process (Scheme 1), SEM and TEM images were utilized to characterize the products of each step. As shown in Fig. 1a, b, the P(VBC-DVB-AA) seed microspheres had relatively smooth surface and narrow particle size distribution of (2.05 ± 0.10) μm and (2.78 ± 0.05) μm, respectively. Due to the selection of the solvent for the supercross-linking reaction, the seed microspheres required a certain degree of cross-linking to maintain their morphology and dispersibility in the subsequent reaction. Meanwhile, the incorporation of the cross-linker DVB also enabled the oligomers of VBC and AA to be successfully precipitated from the mixed solvent of acetonitrile and toluene. Seed microspheres with different particle sizes and crosslinking degrees could be prepared by changing the ratio of monomer VBC and DVB, as shown in Fig. S1. In general, the microsphere particle size gradually increased as the ratio decreases. This may be due to the fact that when the content of DVB was low, some of the oligomers were dissolved in the solvent and cannot be precipitated out [54].

The morphology characterization of different microspheres. a, b SEM images of the PMs-30 and PMs-60; c, d SEM images of the HCPMs-30 and HCPMs-60; e, f SEM and TEM (inset) images of the MHCPMs-30 and the MHCPMs-60; g SEM and particle size distribution (inset) images of the MHCPMs-60 with different magnifications; h SEM and TEM (inset) images of the MPMs; i TEM images of ultrathin sections of the MHCPMs-60; j–l EDX mapping for the ultrathin sections of the MHCPMs-60
The surface of the microspheres was significantly roughened and there was a uniform contrast between light and dark after the hypercrosslinking reaction as shown in Fig. 1d. The polymer chains of PMs were network structure under the action of DVB. Them were fully extended under the swelling of solvent 1, 2-dichloroethane, and were fixed by a large number of methylene bridges formed in the post-crosslinking process. When the solvent was removed at the end of the reaction, the molecular chains were still fixed in a dispersed state, thus forming pores. At this time, the hypercrosslinked microspheres HCPMs-30 and HCPMs-60 still maintained a good spherical shape with narrow particle size distributions of about (2.23 ± 0.05) μm and (2.89 ± 0.10) μm, respectively. Compared with seed microspheres, the particle size of HCPMs increased by about 100 nm to 200 nm, which was consistent with the swelling PMs were fixed by methylene bridges.
The morphology of magnetic hypercrosslinked P(VBC-DVB-AA)@Fe3O4 microspheres was observed by SEM (Fig. 1e, f and h) and TEM (insets). As shown in Fig. 1e, f, magnetic Fe3O4 nanoparticles were prepared by solvothermal method, and in situ grew into Fe3O4 nanoclusters by chelating on HCPMs, which were uniformly distributed on the surface of the microspheres. The Fe3O4 nanoclusters had narrow particle size distribution (about 150 ± 20 nm) and uniform sphericity. Due to the coating of Fe3O4 nanoclusters, the average particle size of the MHCPMs-30 and the MHCPMs-60 increased by about 200 nm to 300 nm over HCPMs. After several hours of reaction at high temperature (about 200℃), HCPMs still maintained regular spherical shape and showed good monodispersity, and no damaged microspheres were observed in a large range of SEM images, which proved the stability of HCPMs. The particle size of the Fe3O4 nanoclusters and their distribution density on the surface of the template microspheres could be controlled by changing the amount of FeCl3 catalyst during the hypercrosslinking process. The appropriate amount of FeCl3 can ensure that the pore structure of the hypercrosslinked microspheres was exposed while the microspheres still had high magnetic responsiveness. Figure 1h is SEM and TEM (inset) images of the MPMs. Under the same reaction conditions, the Fe3O4 nanocluster shells on the MPMs were more compact than those on the MHCPMs. This may be due to the loss of some FeCl3 when the solvent was removed after the hypercrosslinking reaction. It was also possible that the alkaline environment of the solvothermal reaction led to the hydrolysis and oxidation of the chloromethyl groups of the PMs to carboxyl groups, which chelated more Fe3+. For the MPMs, an increase in the concentration of Fe3+ could form dense shell layers on the surface of the PMs (Fig. S2).
In order to further explore the distribution of Fe3O4 nanoparticles inside the magnetic hypercrosslinked P(VBC-DVB-AA)@Fe3O4 microspheres, ultrathin sections of MHCPMs were characterized by TEM. As show in Fig. 1i–l and Fig. S3, it could be seen that the Fe3O4 nanoparticles were uniformly dispersed on the surface of HCPMs in spherical shape. The speculation was further confirmed on the EDX mapping. This indicated that the Fe3O4 nanoparticles were successfully attached to the microsphere surface without clogging the porous structures inside the hypercrosslinked microspheres.
To investigate the components of the materials, FTIR analyses of PMs, HCPMs, MHCPMs and MPMs were performed. Curves of PMs-30 and PMs-60 show the FTIR spectra of PMs obtained with 50% and 20% VBC comonomer addition, respectively. Both curves show C-H stretching vibration peak at 3020 cm−1, C-H stretching vibration peak at 832 cm−1, and C = C stretching vibration peak at 1600, 1500 and 1450 cm−1 on the benzene ring. In addition, the O–H stretching vibration peak at 3448 cm−1, the C = O stretching vibration peak at 1725 cm−1 and the C-O stretching vibration peak at 1167 cm−1 belong to the carboxyl group, a functional group of AA monomer. The presence of C–Cl stretching vibration peak at 676 cm−1 and = C-H in-plane bending vibration peak at 1265 cm−1 confirm the inclusion of the VBC monomer. And compared with the curves of the PMs-30 and the PMs-60, it could be concluded that with the decrease of VBC monomer content, the intensity of the above two peaks also decreased significantly. The above FTIR results confirmed the successful copolymerization of VBC, DVB and AA.
After the Friedel–Crafts alkylation reaction, the characteristic peaks of the chloromethyl groups at 1265 cm−1, 676 cm−1 almost completely disappeared, while the positions of other peaks were substantially unchanged. It indicated that the chloromethyl groups were consumed through hypercrosslinking [55] and the rigid methylene cross-linking bridges were connected in the polymer chains [56]. Subri et al. [55], Zhang et al. [56], and Li et al. [57] reported similar results. Thus, this indicated that hypercrosslinking of P(VBC-DVB-AA) microspheres was successfully carried out.
In the next solvothermal process, iron ions were chelated and adsorbed by the carboxyl functional group of monomer AA, thus forming and assembling Fe3O4 nanoclusters on the template microspheres. In curves of the MHCPMs-60 and the MPMs, the Fe–O stretching vibration peak at 580 cm−1 confirms the formation of Fe3O4. In summary, magnetic hypercrosslinked P(VBC-DVB-AA)@Fe3O4 microspheres and magnetic P(VBC-DVB-AA)@Fe3O4 microspheres have been successfully synthesized.
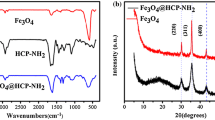
XRD characterization of the MPMs, the MHCPMs-30 and the MHCPMs-60 was performed to determine the crystal form of the Fe3O4 nanoparticles. The diffraction peaks of the three synthesized magnetic microspheres were consistent with those of typical magnetite, as shown in Fig. 2b. The diffraction peaks at 2θ = 30.12°, 35.48°, 43.12°, 53.50°, 57.03° and 62.63° were attributed to the crystalline planes (220), (311), (400), (422), (511) and (440) of Fe3O4, respectively. These results indicated that the Fe3O4 nanoparticles with cubic spinel structure were successfully synthesized [58]. In addition, the diffraction peak intensity of each sample was also related to the density of Fe3O4 nanoclusters on the surface of the microspheres, which was consistent with the SEM results.

a FT-IR spectra of PMs, HCPMs, MHCPMs and the MPMs; b XRD patterns of the MPMs, the MHCPMs-30 and the MHCPMs-60; c TGA curves of the PMs-30, the MPMs, the HCPMs-30, the MHCPMs-30 and the MHCPMs-60; d Magnetization curves of the MPMs, the MHCPMs-30 and MHCPMs-60; and the inset is the magnifying scale of the plot
The thermal stability of the PMs-30, HCPMs-30, MHCPMs-30 and MHCPMS-60 was characterized by thermogravimetric analysis (TGA) under N2 atmosphere, and the curves are shown in Fig. 2c. At the initial stage, all four samples showed a slight mass loss due to the evaporation of water adsorbed by the hydrophilic groups and porous structure. And the weight loss of the four samples at this stage was about 10%. Subsequently, significant weight loss began to occur from about 300 °C to 500 °C, which was thought to be due to decomposition of the polymer organic skeleton. At this time, the HCPMs-30 and MHCPMs still had more than 40% remaining mass compared to crosslinked seed microspheres. After 550 °C, the weight loss of the hypercrosslinked microspheres and magnetic microspheres appeared in the third stage, which may be related to the hypercrosslinking process. It may be due to the final decomposition of crosslinked or hypercrosslinked aromatic groups. The final residual mass ratio of the PMs-30 and HCPMs-30 was 11.4% and 31.5%, respectively, indicating that the thermal stability of the microspheres was significantly improved by the hypercrosslinking process. In addition, the residual mass ratios of the MPMs, MHCPMs-30 and MHCPMs-60 were 31.6%, 43.5% and 43.9%, respectively. Based on these data, the weight percentage of Fe3O4 in the MPMs, MHCPMs-30 and MHCPMs-60 was estimated to be 20.1%, 12.0% and 12.4%, respectively.
The magnetic behaviors of the MPMs, the MHCPMs-30 and the MHCPMs-60 were also measured by using a vibrating sample magnetometer. Comparison of the three curves in Fig. 2d shows that the saturation magnetization intensity value of the MPMs was 46.3 emu/g, which was higher than that of the MHCPMs-60 (Ms = 29.0 emu/g) and the MHCPMs-30 (Ms = 23.7 emu/g). This phenomenon may be due to the higher concentration of Fe3+ in the PMs’ system under the same reaction conditions. Combined with TEM images (Fig. 1), it can be seen that Fe3O4 nanoclusters on the MPMs surface were more densely arranged and had larger particle sizes. At the same time, the local TEM and electron diffraction images of the MPMs (Fig. S4) shows that Fe3O4 nanoclusters had grown into single crystal particles. Therefore, the MPMs had a certain degree of coercivity and exhibited paramagnetism. In contrast, the Fe3O4 nanoclusters of the MHCPMs-30 and the MHCPMs-60 had rough surfaces and were composed of many small size Fe3O4 nanoparticles, which were polycrystalline. This made the Fe3O4 nanoclusters of MHCPMs not only have the paramagnetic properties of Fe3O4 nanoparticles, but also acquire stronger magnetic properties due to the aggregation of Fe3O4 nanoparticles. And the hysteresis loops also proved that MHCPMs exhibited near-zero coercivities and remanence. As a result, these microspheres can be easily magnetically separated (Fig. 3), which is a major advantage of magnetic adsorbents in wastewater treatment.

a magnetic separation photographs of the MHCPMs-60; b magnetic separation photographs of the MHCPMs-60 after adsorption of dye
The N2 sorption isotherms and pore size distribution of the HCPMs-1, HCPMs-4, MHCPMs-30, and MHCPMs-60 are presented in Fig. 4. According to the IUPAC[59] classification method, all the above samples were typically characterized by type-I classification. When the relative pressure was less than 0.1, there was a rapid rising phase in the curves of all samples, indicating that they had a well-developed microporous structure. With the increase of relative pressure, the adsorption increased slowly, and when the relative pressure was between 0.4 and 1, a hysteresis loop could be observed, which was most obvious for the HCPMs-30, indicating the presence of mesoporous structures in the microspheres. Combined with the pore size distribution diagram, it can be proved that the pore structures of the microspheres were mainly microporous structures, and also contained some mesoporous structures with a diameter of about 2 ~ 4 nm. In addition, Fig. S5 showed that the N2 adsorption isotherms of PMs were close to type-III classification, indicating that the PMs-30 and the PMs-60 may be nonporous or macroporous solid materials. Comparing the N2 adsorption isotherms of PMs and HCPMs, the N2 adsorption capacity of HCPMs were much larger than that of PMs under the same relative pressure, which suggested that HCPMs have larger specific surface areas. This may be attributed to the formation of a large number of micropores through methylene crosslinking bridges during the Friedel–Crafts reaction, indicating the successful conduct of hypercrosslinking.

a, b N2 adsorption–desorption isotherms and the mesopore pore size distribution (inset) of the HCPMs-30 and the HCPMs-60; c, d N2 adsorption–desorption isotherms and the mesopore pore size distribution (inset) of the MHCPMs-30 and the MHCPMs-60
The specific surface area, the pore volume, and the average pore diameter of HCPMs and MHCPMs calculated using standard procedures from N2 adsorption isotherms are given in Table 4. The specific surface areas of the PMs-30 and the PMs-60 were merely 4.21 m2/g and 7.31 m2/g, which may be due to the formation of pores by particle stack or the accumulation of N2 at the most attractive parts of its surface. This was consistent with the N2 adsorption isotherm results of the PMs above. Compared with the seed microspheres of the PMs, the results indicated that the hypercrosslinking reaction produced extensive porosity in the HCPMs by generating numerous crosslinked bridges. In addition, the microporous area of the HCPMs gradually increased with the increase of DVB concentration in the monomer mixtures, whereas the average pore size gradually decreased. When the microporous surface area reached the maximum value of 658.74 m2/g, the specific surface area of the microspheres also reached a relatively high value of 890.47 m2/g. This may be attributed to the fact that DVB acts as a crosslinking agent to provide the microspheres with a preliminary crosslinking immobilization network during the copolymerization process. In the hypercrosslinking process, when the content of DVB was low, the chloromethyl functional groups on the benzene ring were more likely to contact with the molecular chains of the benzene rings that were far away from each other, and larger pore structures were formed; when the content of DVB was high, the molecular chains of the microspheres can only be in the stretching state, and the benzene rings can only be crosslinked with the neighboring benzene rings, so that the denser crosslinked network was obtained in the end, and the size of pore size decreased in comparison [60].
The specific surface area and pore structure of adsorbents play an important role in the adsorption process, and the HCPMs-30 and the HCPMs-60 possessed higher specific surface areas, so they were chosen to the synergistic fabrication of magnetic porous microspheres (the MHCPMs-30 and the MHCPMs-60). And their N2 adsorption isotherms and pore size distributions are shown in Fig. 4c, d, respectively. Compared with the hypercrosslinked microspheres, the surface area of the magnetic microspheres decreased, but still remained above 600 m2/g. The pore volume also decreased, but the average pore size increased slightly, which may be due to the blockage of some of the micropores by Fe3O4 nanoparticles. Combined with the TEM images of ultrathin sections of the MHCPMs, it can be seen that the Fe3O4 nanoclusters chelated on the porous microspheres only covered a part of the pore structures on the surface of the microspheres and no obvious magnetic particles were observed inside the microspheres. Therefore, MHCPMs still retained considerable surface areas and pore volumes.
9 Adsorption of MB on MHCPMs
9.1 Effect of the Initial pH Value
After the detailed characterization, the prepared MHCPMs had the characteristics of large specific surface area, strong magnetic properties and surface functionalization. The microspheres were rich in carboxyl functional groups, which can adsorb cationic dye (MB) in water by electrostatic interaction. Therefore, the pH of the MB solution was critical to the adsorption capacity of adsorbents due to the charge distribution. The adsorption performance of the adsorbent under different conditions was analyzed using UV–Vis spectrophotometer.
Firstly, the effect of pH on the adsorption performance of the MHCPMs-60 was investigated. Using 40 mL of 15 mg/L MB solution, the pH was adjusted to 2–12 with 0.1 mol/L HCl or 0.1 mol/L NaOH solution, and added 4 mg of the adsorbent. Figure 5a shows that the dye removal rate increased significantly with increasing pH. To elucidate this effect, the zeta potential of the MHCPMs-60 microspheres was measured at different pH. The results indicated that the zeta potential of the microspheres showed a negative to positive trend with increasing pH, which could be attributed to the presence of carboxyl functional groups in the MHCPMs-60. Alkaline conditions would promote the ionization of the carboxyl group, resulting in the microspheres acquiring more negative charge. Thus, the electrostatic adsorption effect of the microspheres on cationic dyes was stronger. Under acidic conditions, the concentration of H+ was higher, which may inhibit the ionization of the carboxyl groups or form competitive adsorption with cationic dyes on the microsphere surface. This led to a decrease in the adsorption capacity of the microspheres for dyes. However, even at the pH of 2, the removal rate of MB remained at 78%, indicating that the adsorption of dyes by microspheres was not only controlled by the electrostatic interaction. When the pH value of the solution was 7, the removal rate of MB from MHCPMs-60 was still as high as 96.3%. Therefore, in order to avoid secondary pollution, neutral conditions were selected for follow-up experiments.

a effect of pH for MB adsorption (C0 = 20 mg/L); b effect of different initial methylene blue solution concentration on the adsorption of the MPMs, MHCPMs-30 and MHCPMs-60 (T = 25℃, pH = 7); c effect of contact time for MB adsorption (pH = 7, T = 25℃, C0 = 20 mg/L); d reusability of the MHCPMs-60 for MB adsorption (pH = 7, T = 25℃, C0 = 10 mg/L). The error bars indicate the standard deviation from the mean (n = 3)
9.2 Effect of the Initial MB Concentration
Subsequently, the adsorption properties of the MPMs, MHCPMs-30 and MHCPMs -60 in aqueous neutral methylene blue solutions with different initial concentrations were studied separately. Figure 5b shows the effect of different initial concentrations of methylene blue solutions on the adsorption capacity. The MB adsorption on the MHCPMs-60 increased from 49.9 to 214.26 mg/g as the initial concentration was increased from 5 to 50 mg/L. The MHCPMs-30, which possesses similar specific surface area to the MHCPMs-60, only increased its MB adsorption from 49.9 to 156.4 mg/g. Combined with Table 1, it can be seen that the average pore sizes of both the MHCPMs-30 and MHCPMs-60 were around 2–3 nm, while the particle size of the MHCPMs-60 was larger. Therefore, the combined consideration of the size of the dye molecules and the steric effects in the adsorption process ultimately led to the higher adsorption capacity of the MHCPMs-60. The same adsorption was observed on the MPMs without hypercrosslinking, but the MB adsorption increased from 43.5 to 80.7 mg/g, which was much lower than that on the MHCPMs. In contrast, the adsorption of MB by the MPMs was driven by hydrogen bonding and electrostatic interactions between the microspheres and MB, whereas the source of adsorption driving force for the MHCPMs was also coupled with their rich porous structure.
9.3 Effect of Adsorption Temperature
Under the conditions of 25–45 °C, 4 mg of magnetic microspheres were added to 40 mL of 5–50 mg/L methylene blue solution, and the effect of different temperatures on the adsorption of MB solution was investigated. As show in Fig. S6, the equilibrium adsorption amount of the dye decreased when the adsorption temperature was increased from 25 °C to 45 °C, and the dye removal rate also decreased. The experiments indicated that the low temperature was more favorable for the adsorption process.
9.4 Effect of Adsorption Time
As the results shown in Fig. 5C, the adsorption capacity of the MHCPMs-30 and MHCPMs-60 initially increased with the increase in time and then gradually remained unchanged. More than 90% of the dye was removed within 30 min and the reaction reached equilibrium within 60 min. Based on the slope of the curves, the samples exhibited a fast removal rate for MB in the initial stage, and up to 73% of MB was removed in the first 5 min. As the reaction time was extended further, the rate of adsorption slowed down significantly. This was in line with the property of microporous materials to reach adsorption equilibrium quickly.
9.5 Adsorption Isotherms
To further understand the adsorption process and evaluate the adsorption capacity of the MHCPMs-30and MHCPMs-60 on MB more comprehensively, the Langmuir and Freundlich models were applied to analyze the adsorption equilibrium process characteristics at 25℃ and pH = 7. The linearized Langmuir and Freundlich adsorption isotherm equations were:
Langmuir equation:
Freundlich equation:
where the Ce (mg/L) was represented for the equilibrium adsorption concentration of MB; the Qe (mg/g) was the equilibrium sorption capacity of adsorbent; Qm (mg/g) was the theoretical maximum adsorption capacity of adsorbent; KL(L/mg) was the Langmuir equilibrium constant; KF (mg1−nLn/g) was the Freundlich equilibrium constant; 1/n was the adsorption degree.
The equilibrium curves of the Langmuir and Freundlich isotherms for the two adsorbents are depicted in Fig. 6a, b, and the relevant parameters are given in Table 4. It could be seen that the Langmuir model well described the MB adsorption process for both adsorbents, in particular, the correlation coefficient for the MHCPMs-60 was 0.9999 (0.9168 for Freundlich isotherm). This indicated that the adsorption process was homogeneous and monolayer in the concentration range studied [61]. The value of the maximum adsorption capacity (Qm) calculated from the Langmuir model for the MHCPMs-30 was 215.52 mg/g, which was consistent with the experimental value (214.26 mg/g). In addition, the n value of the Freundlich adsorption model was greater than one, which indicated that MHCPMs had better adsorption performance and great MB removal potential (Table 5).

a Langmuir isotherms and b Freundlich isotherms for MB adsorption (pH = 7, T = 25℃); c pseudo-first-order plots and d pseudo-second-order plots for MB adsorption (pH = 7, T = 25℃, C0 = 20 mg/L)
9.6 Adsorption Kinetics Study
In order to understand the adsorption process of methyl bromide by the MHCPMs-30 and the MHCPMs-60 composites, pseudo-first-order kinetic model and pseudo-second-order kinetic model[62] were used to simulate the experimental data at 25℃ and pH = 7. The equations of the above two models were expressed as follows:
where Qe (mg/g) was the adsorption capacity at equilibrium time, Qt (mg/g) was the adsorption capacity at any time, k1 (min−1) and k2 (g/mg·min) were the pseudo-first order rate constant and pseudo-second-order rate constant, respectively.
The results of the model fitting are shown in Fig. 6c, d, and the kinetic parameters and correlation coefficient values R2 determined by linear regression are listed in Table 6. Pseudo-first-order equation better described the adsorption kinetics of MB onto the MHCPMs-30 and MHCPMs-60 composites than pseudo-second-order equation. In addition, the Qe values of the pseudo-first-order model were very close to the experimental values. Hence, the pseudo-first-order kinetic model was suitable to describe the rate of MB adsorption by MHCPMs at concentration studied. The above results indicated that the external diffusion was a significant step in adsorption, and that the process was pore-filling or physical adsorption [63]. In addition, dye molecules may form imaginary bonds as a result of the mutual interactions with active sites on MHCPMs via electrostatic interaction, and van der Waals and dispersive forces [64]. For instance, electron transfer occurred between the carboxyl group of MHCPMs and the nitrogen atom in MB, forming an adsorption chemical bond [47]. It is also suggested that these bonds are relatively weak, and the adsorption is therefore reversible.
9.7 Recycle Performance of the MHCPMs-60
As shown in Fig. 5d, the recyclability of the MHCPMs-60 was investigated. It can be seen that the adsorption capacity of the MHCPMs-60 gradually decreased with the increase of the number of cycles. This may be due to the presence of carboxyl groups in the microspheres, which form chemical bonds with the dye molecules, resulting in the dye difficult to completely remove during elution. However, the adsorption capacity of the prepared microspheres mainly depended on the rich pore structures, so the dye adsorption capacity still remained at a high level. After seven consecutive adsorption–desorption cycles at the concentration of MB solution of 10 mg/L, the dye removal rate of the MHCPMs-60 was still above 90%. This indicated that the MHCPMs-60 exhibited good stability and recyclability.
Above all, we could draw the conclusion that the adsorption process of MB was a Langmuir process and the adsorption behavior belonged to the pseudo-first-order model. The adsorption behavior of MHCPMs on MB was mainly related to their abundant porous structure, hydrogen bonding interactions, electrostatic interactions and chemical sorption between the MHCPMs and MB. The introduction of carboxyl functional groups can provide more adsorption sites on the surface of the porous channel. Moreover, the abundant porous structures further improved the accessibility of the adsorption sites and accelerated the full adsorption. The comparison of the maximum MB adsorption capacity of the MHCPMs prepared in this paper with other adsorbents reported in recent literature is shown in Table 7. The preparation of MHCPMs made secondary use of the catalyst to obtain magnetic porous microspheres, which was not only environmentally friendly, but also achieved the effect of rapid separation. At the same time, MHCPMs also had excellent adsorption and cycling properties. Therefore, the MHCPMs were a more highly efficient adsorbent for the removal of MB than several other sorption materials, which had a great potential application in water treatment.
10 Conclusions
In summary, this work proposed a synergistic and facile method for preparing magnetic hypercrosslinked porous polymer microspheres. Friedel–Crafts alkylation reaction of P (VBC-DVB-AA) microspheres with different crosslinking degrees was carried out with FeCl3 as catalyst to obtain hypercrosslinked porous microspheres. Without any washing operation, the catalyst in the previous step was used as the iron source of the solvothermal reaction, and Fe3O4 nanoclusters were in-situ generated on the surface of the hypercrosslinked microspheres to obtain MHCPMs. In this method, the catalyst was reused, which saved resources and avoids the problem that the products of hypercrosslinking reaction are difficult to clean. Simultaneously, the use of polymer microspheres with different crosslinking degrees as seed microspheres not only regulated the pore size structures of the hypercrosslinked microspheres, but also greatly improved the rigidity and solvent resistance of the microspheres. And the monomer AA was introduced into the seed microspheres to provide rich functional groups for the hypercrosslinked microspheres. MHCPMs were characterized by SEM, TEM, EDX, XRD, FTIR, TGA, VSM and BET. The characterization results indicated that the MHCPMs-60 had the characteristics of uniform particle size, high specific surface area (654.56m2/g), superparamagnetism and a high saturation magnetization (29.0 emu/g). According to the characteristics of this microspheres, it was applied to remove cationic dyes from water. The experimental results indicated that MHCPMs can quickly and effectively remove methylene blue dye from water. They had a high specific surface area and abundant pore structure, and MB can be transferred from the surface to the internal micropores through pore channels. The maximum adsorption capacity of the MHCPMs-60 was 214.26 mg/g and the maximum dye removal rate was 99.9%. The adsorption kinetics fit the pseudo-first-order model, and the isothermal adsorption equilibrium data fit the Langmuir model. In addition, the MHCPMs-60 maintained a good adsorption capacity after 7 cycles of adsorption. Hence, the strategy applied for efficient water treatment by magnetic hypercrosslinked porous P(VBC-DVB-AA)@Fe3O4 microspheres is promising for practical applications.
Data Availability
No datasets were generated or analysed during the current study.
References
R. Noor, A. Maqsood, A. Baig, C.B. Pande, S.M. Zahra, A. Saad, M. Anwar, S.K. Singh, Urban Clim. 48, 101413 (2023). https://doi.org/10.1016/j.uclim.2023.101413
L. Zhang, Z. Xin, X. Fei, H. Luo, H. Li, B. Lu, Z. Li, G. Wei, J. Water Supply Res. Technol-Aqua. 68(1), 39–50 (2019). https://doi.org/10.2166/aqua.2018.100
J.D. Sharpe, J.A. Kaufman, Z.E. Goldman, A. Wolkin, M.O. Gribble, Environ. Res. 170, 472–480 (2019). https://doi.org/10.1016/j.envres.2019.01.009
D. Huang, L. Gao, S. Zhu, L. Qiao, Y. Liu, Q. Ai, C. Xu, W. Wang, M. Lu, M. Zheng, Chemosphere 324, 138314 (2023). https://doi.org/10.1016/j.chemosphere.2023.138314
L. Jin, Y. Wang, T. Xue, J. Xie, Y. Xu, Y. Yao, X. Li, Langmuir 35(45), 14473–14480 (2019). https://doi.org/10.1021/acs.langmuir.9b02583
M.L. Yola, Chemosphere 301, 134766 (2022). https://doi.org/10.1016/j.chemosphere.2022.134766
O. Moradi, I.D. Sharabaf, Environ. Res. 214, 114006 (2022). https://doi.org/10.1016/j.envres.2022.114006
S. Yadav, A. Asthana, A.K. Singh, R. Chakraborty, S.S. Vidya, M.A.B.H. Susan, S.A.C. Carabineiro, J. Hazard. Mater. 409, 124840 (2021). https://doi.org/10.1016/j.jhazmat.2020.124840
H. Zheng, X. Yang, K. Meng, S. Li, H. Yu, Q. Peng, Y. Zhang, X. Zhang, X. Xu, Y. Zhang, Z. Xu, L. Li, Q. Ying, N.I. Elsheery, J. Soil Sci. Plant Nutr. 23(2), 2599–2609 (2023). https://doi.org/10.1007/s42729-023-01216-4
Y. Chai, D.J. Pannell, P.G. Pardey, Food Policy 120, 102525 (2023). https://doi.org/10.1016/j.foodpol.2023.102525
J. Fořt, K. Kobetičová, M. Böhm, J. Podlesný, V. Jelínková, M. Vachtlová, F. Bureš, R. Černý, Polymers 14(7), 1416 (2022). https://doi.org/10.3390/polym14071416
M.B. McGechan, A. Barnes, R. Fychan, C.L. Marley, Comput. Electron. Agric. 142, 397–405 (2017). https://doi.org/10.1016/j.compag.2017.09.022
B. Bharathiraja, I.A. Ebenezer Selvakumari, J. Iyyappan, S. Varjani, Curr. Opin. Environ. Sci. Health. 12, 6–17 (2019). https://doi.org/10.1016/j.coesh.2019.07.004
D.A. Yaseen, M. Scholz, Int. J. Environ. Sci. Technol. 16(2), 1193–1226 (2018). https://doi.org/10.1007/s13762-018-2130-z
E. Ugochukwu, O. D. Saliu, K. Dulta, S. Ogunniyi, A. O. Bajeh, K. O. Iwuozor, & J. O. Ighalo, Water Process Eng. 50, 103273 (2022). https://doi.org/10.1016/j.jwpe.2022.103273
Y. Wu, M. Chen, H.-J. Lee, M.A. Ganzoury, N. Zhang, C.-F. de Lannoy, ACS ES&T Eng. 2(9), 1574–1598 (2022). https://doi.org/10.1021/acsestengg.2c00201
Y. Hao, Z. Wang, Z. Wang, Y. He, Ecotoxicol. Environ. Saf. 168, 298–303 (2019). https://doi.org/10.1016/j.ecoenv.2018.10.076
O. León, A. Muñoz-Bonilla, D. Soto, D. Pérez, M. Rangel, M. Colina, M. Fernández-García, Carbohydr. Polym. 194, 375–383 (2018). https://doi.org/10.1016/j.carbpol.2018.04.072
L. Wang, A. Wang, Bioresour. Technol. 99(5), 1403–1408 (2008). https://doi.org/10.1016/j.biortech.2007.01.063
Z.-L. Cheng, Y.-X. Li, Z. Liu, Ecotoxicol. Environ. Saf. 148, 585–592 (2018). https://doi.org/10.1016/j.ecoenv.2017.11.005
H. Zhang, Y. Li, B. Cheng, C. Ding, Y. Zhang, Int. J. Biol. Macromol. 161, 561–572 (2020). https://doi.org/10.1016/j.ijbiomac.2020.06.017
B. Marta, T.P. Gemma, P.C. Carlos, Sep. Purif. Technol. 303, 122211 (2022). https://doi.org/10.1016/j.seppur.2022.122211
S.H. Teo, C.H. Ng, A. Islam, G. Abdulkareem-Alsultan, C.G. Joseph, J. Janaun, Y.H. Taufiq-Yap, S. Khandaker, G.J. Islam, H. Znad, M.R. Awual, J. Cleaner Prod. 332, 130039 (2022). https://doi.org/10.1016/j.jclepro.2021.130039
Y. Zhu, W. Fan, T. Zhou, X. Li, Sci. Total. Environ. 678, 253–266 (2019). https://doi.org/10.1016/j.scitotenv.2019.04.416
M. Naushad, A.A. Alqadami, A.A. Al-Kahtani, T. Ahamad, M.R. Awual, T. Tatarchuk, J. Mol. Liq. 296, 112075 (2019). https://doi.org/10.1016/j.molliq.2019.112075
D. Wu, F. Xu, B. Sun, R. Fu, H. He, K. Matyjaszewski, Chem. Rev. 112(7), 3959–4015 (2012). https://doi.org/10.1021/cr200440z
N. Fontanals, R.M. Marcé, F. Borrull, P.A.G. Cormack, Polym. Chem. 6(41), 7231–7244 (2015). https://doi.org/10.1039/c5py00771b
P.M. Budd, G.B.S., M. Saad, N.B. McKeown, K.J. Msayib, C.E. Tattershall, Chem. Commun. 2, 230–231 (2004). https://doi.org/10.1039/B311764B
A.I. Cooper, Adv. Mater. 21(12), 1291–1295 (2009). https://doi.org/10.1002/adma.200801971
S.-Y. Ding, W. Wang, Chem. Soc. Rev. 42(2), 548–568 (2013). https://doi.org/10.1039/c2cs35072f
D. Robert, D.J. Adams, A.I. Cooper, Chem. Sci. 2(6), 1173 (2011). https://doi.org/10.1039/c1sc00100k
C.E. Chan-Thaw, A. Villa, P. Katekomol, D. Su, A. Thomas, L. Prati, Nano Lett. 10(2), 537–541 (2010). https://doi.org/10.1021/nl904082k
R. Anuj, M. Raeesh, R.K. Singh, J. Pratiksha, O.P. Khatri, S.V. Chandra, M. Paritosh, J. Cleaner Prod. 418, 138188 (2023). https://doi.org/10.1016/j.jclepro.2023.138188
Y. Li, S. Zheng, X. Liu, P. Li, L. Sun, R. Yang, S. Wang, Z.S. Wu, X. Bao, W.Q. Deng, Angew. Chem. Int. Ed. 57(27), 7992–7996 (2017). https://doi.org/10.1002/anie.201711169
Z. Chuntian, D. Ekram, C. N. R. and K. Ritu, J. Mater. Chem. 17, 23, 2446 (2007). https://doi.org/10.1039/b700929a
R. Dawson, A.I. Cooper, D.J. Adams, Prog. Polym. Sci. 37(4), 530–563 (2012). https://doi.org/10.1016/j.progpolymsci.2011.09.002
N. Liu, J. Chen, Z. Wu, P. Zhan, L. Zhang, Q. Wei, F. Wang, L. Shao, A.C.S. Appl, Polym. Mater. 3(4), 2178–2188 (2021). https://doi.org/10.1021/acsapm.1c00139
P. Shi, X. Chen, Z. Sun, C. Li, Z. Xu, X. Jiang, B. Jiang, J. Colloid Interface Sci. 563, 272–280 (2020). https://doi.org/10.1016/j.jcis.2019.12.038
Q. Li, Z. Zhan, S. Jin, B. Tan, Chem. Eng. J. 326, 109–116 (2017). https://doi.org/10.1016/j.cej.2017.05.049
L. Ziwang, W. Junmin, G. Yaxing, L. Jiajia, W. Juntao, W. Chun, W. Qiuhua, W. Zhi, J. Chromatogr. A 1676, 463206 (2022). https://doi.org/10.1016/j.chroma.2022.463206
H. Man, Y. Nie, S. Shao, Y. Wang, Z. Wang, Y. Jiang, New J. Chem. 46(28), 13442–13453 (2022). https://doi.org/10.1039/d2nj01440h
C. Bo, C. Yangrui, Z. Huinan, L. Fengxia, F. Xiang, L. Juan, P. Xuejun, J. Hazard. Mater. 392, 122263 (2020). https://doi.org/10.1016/j.jhazmat.2020.122263
S. Jian, L. Lijie, Y. Rui, M. Xianlong, J. Shaohua, C. Kun, C. Shusen, L. Xijuan, S. Qinghai, Molecules 25(13), 3044 (2020). https://doi.org/10.3390/molecules25133044
J. Ge, Y. Hu, M. Biasini, W.P. Beyermann, Y. Yin, Angew. Chem. 119(23), 4420–4423 (2007). https://doi.org/10.1002/ange.200700197
S.E. Shirsath, D. Wang, S.S. Jadhav, M.L. Mane, S. Li, Ferrites Obtained by Sol-Gel Method (Springer International Publishing, Cham, 2018), pp. 695–735
H. Deng, X. Li, Q. Peng, X. Wang, J. Chen, Y. Li, Angew. Chem. Int. Ed. 44(18), 2782–2785 (2005). https://doi.org/10.1002/anie.200462551
Z. Jiaqi, D. Yimin, L. Danyang, W. Shengyun, Z. Liling, & Z. Yi, Colloids Surf. A. 572, 58–66 (2019). https://doi.org/10.1016/j.colsurfa.2019.03.095
G. Wang, Q. Liu, M. Chang, J. Jang, W. Sui, C. Si, Y. Ni, Ind. Crops Prod. 127, 110–118 (2019). https://doi.org/10.1016/j.indcrop.2018.10.056
T. Yao, S. Guo, C. Zeng, C. Wang, L. Zhang, J. Hazard. Mater. 292, 90–97 (2015). https://doi.org/10.1016/j.jhazmat.2015.03.014
T. Xue, F. Shao, H. Miao, X. Li, Environ. Sci. Pollut. Res. 30(43), 97147–97159 (2023). https://doi.org/10.1007/s11356-023-29102-7
J. Jin, T. Feng, Y. Ma, W. Wang, Y. Wang, Q. Zhou, A. Li, Chemosphere 185, 563–573 (2017). https://doi.org/10.1016/j.chemosphere.2017.07.058
X. Zou, Z. Wei, J. Du, X. Wang, G. Zhang, Chem. Res. Chin. Univ. 34(4), 684–690 (2018). https://doi.org/10.1007/s40242-018-7413-x
N. Fontanals, R.M. Marcé, P.A.G. Cormack, D.C. Sherrington, F. Borrull, J. Chromatogr. A 1191(1–2), 118–124 (2008). https://doi.org/10.1016/j.chroma.2007.11.061
S.E. Shim, S. Yang, H.H. Choi, S. Choe, J. Polym. Sci. Part A: Polym. Chem. 42(4), 835–845 (2004). https://doi.org/10.1002/pola.11028
N.N.S. Subri, P.A.G. Cormack, S.N.AMd. Jamil, L.C. Abdullah, R. Daik, J. Appl. Polym. Sci. 135, 2 (2017). https://doi.org/10.1002/app.45677
T. Zhang, J. Huang, J. Colloid Interface Sci. 505, 383–391 (2017). https://doi.org/10.1016/j.jcis.2017.06.030
X. Li, Y. Liu, D. Di, G. Wang, Y. Liu, Colloids Surf. A. 500, 1–9 (2016). https://doi.org/10.1016/j.colsurfa.2016.03.061
Y. Wang, Y. Wei, P. Gao, S. Sun, Q. Du, Z. Wang, Y. Jiang, A.C.S. Appl, Mater. Interfaces. 13(9), 11166–11176 (2021). https://doi.org/10.1021/acsami.0c19734
M. Thommes, K. Kaneko, A.V. Neimark, J.P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol, K.S.W. Sing, Pure Appl. Chem. 87(9–10), 1051–1069 (2015). https://doi.org/10.1515/pac-2014-1117
L. Tan, B. Tan, Chem. Soc. Rev. 46(11), 3322–3356 (2017). https://doi.org/10.1039/c6cs00851h
I. Langmuir, J. Am. Chem. Soc. 40(9), 1361–1403 (1918). https://doi.org/10.1021/ja02242a004
G.M.Y.S. Ho, Process Biochem. 34, 451–465 (1999). https://doi.org/10.1016/S0032-9592(98)00112-5
A.N.M. Faizal, M.H.A. Halim, M.A.A. Zaini, Carbon Lett. 29(4), 319–326 (2019). https://doi.org/10.1007/s42823-019-00046-8
É.C. Lima, M.A. Adebayo, F. M. Machado, Carbon Nanomaterials as Adsorbents for Environmental and Biological Applications, ed. By C.P. Bergmann, F.M. Machado (Springer International Publishing, Cham, 2015), pp. 33–69
Y. He, W. Bao, Y. Hua, Z. Guo, X. Fu, B. Na, D. Yuan, C. Peng, H. Liu, RSC Adv. 12(9), 5587–5594 (2022). https://doi.org/10.1039/d1ra08589a
L.-W. Jiang, F.-T. Zeng, Y. Zhang, M.-Y. Xu, Z.-W. Xie, H.-Y. Wang, Y.-X. Wu, F.-A. He, H.-L. Jiang, Adv. Powder Technol. 32(2), 492–503 (2021). https://doi.org/10.1016/j.apt.2020.12.026
S. Liu, X. Chen, W. Ai, C. Wei, J. Cleaner Prod. 212, 1062–1071 (2019). https://doi.org/10.1016/j.jclepro.2018.12.060
K. Zhou, Y. Li, Q. Li, Q. Du, D. Wang, K. Sui, C. Wang, H. Li, Y. Xia, J. Polym. Environ. 26(8), 3362–3370 (2018). https://doi.org/10.1007/s10924-018-1219-2
G. Xiong, S. Gao, Q. Zhang, B. Ren, L. You, F. Ding, Y. He, Y. Sun, Polymer 247, 124787 (2022). https://doi.org/10.1016/j.polymer.2022.124787
J. Song, L. Chen, Y. Niu, Z. Wei, Y. Sun, J. Polym. Environ. 31(7), 2999–3014 (2023). https://doi.org/10.1007/s10924-023-02781-6
M. Zhou, T. Wang, Z. He, Y. Xu, W. Yu, B. Shi, K. Huang, ACS Sustainable Chem. Eng. 7(3), 2924–2932 (2019). https://doi.org/10.1021/acssuschemeng.8b01807
J. Zhang, H. Man, X. Han, Z. Wang, Y. Jiang, Colloid Polym. Sci. 301(8), 933–947 (2023). https://doi.org/10.1007/s00396-023-05099-w
B. Yu, L. He, Y. Wang, H. Cong, Materials. 10(11), 1239–1251 (2017). https://doi.org/10.3390/ma10111239
Acknowledgements
This work was supported by the Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) and the Fundamental Research Funds for the Central Universities with grant number 2242016K41020.
Author information
Authors and Affiliations
Contributions
DW: conceptualization, investigation, data curation, and writing – original draft. QX: writing – review & editing and supervision. YN: validation and formal analysis. WJ: investigation. ZW: supervision, conceptualization, writing – review & editing, and funding acquisition. JY: supervision, conceptualization, writing – review & editing, and funding acquisition.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wu, D., Xu, Q., Nie, Y. et al. Synergistic Fabrication of Magnetic Hypercrosslinked Porous P(VBC-DVB-AA)@Fe3O4 Microspheres for Removal of Cationic Dye. J Inorg Organomet Polym 34, 2746–2761 (2024). https://doi.org/10.1007/s10904-024-03009-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10904-024-03009-3