
Abstract
Two new isomorphous zinc(II) and nickel(II) supramolecular complexes, [Zn(L)2(H2O)2]n, (1), and [Ni(L)2(H2O)2]n (2) {HL = (benzotriazol-1-yloxy)-acetic acid} have been synthesized under ambient conditions by the self-assembly of HL with Zn(Ac)2·2H2O or Ni(Ac)2·4H2O in the presence of triethylamine. The two complexes have been characterized by single-crystal X-ray diffraction, elemental analysis, and IR spectroscopy. Single-crystal X-ray diffraction shows that the two complexes are isostructural and crystallize in monoclinic crystal system P21/c with similar unit-cell parameters and exhibit one-dimensional infinite chain structures. Adjacent chains are linked by O–H⋯O hydrogen bonds, giving rise to a two-dimensional network and these networks are further extended by weak π⋯π stacking interactions among adjacent aromatic cycles of HL ligands into a three-dimensional framework. In addition, the luminescent property of 1 and UV–Visible absorbance of 2 were also investigated and discussed. Density functional theory (DFT) calculations of the structures, stabilities, orbital energies, and composition characteristics of some frontier molecular orbitals of the two complexes were performed by means of Gaussian 09W package and taking B3LYP basis set.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Coordination polymers as a new kind of inorganic–organic hybrid materials have attracted great interest in recent years due to their interesting aesthetic structures and topologies [1], and potential applications in many fields, such as gas storage and separation [2], nonlinear optics [3, 4], magnetism [5, 6], luminescence [7], sensing [8, 9], catalysis [10, 11], energy storage [12,13,14,15,16,17,18,19], and so on. The design and synthesis of such materials are influenced by many factors such as the selection of metal ions, the structural characteristics of the organic ligand, the solvent, pH, and the other reaction conditions [20,21,22,23]. Among these factors, the selection of appropriate ligand plays a key role in the formation of final structures of coordination polymers. The combination of neutral N-donor compounds and carboxylic acids has been considered as an effective strategy for constructing interesting coordination polymers in recent years [24,25,26]. But supramolecular complexes based on semi-rigid organic ligands containing 1-hydroxybenzotriazole and carboxylic acids have remained rare until recently. Up to now, only one complex constructed from (benzotriazol-1-yloxy)-acetic acid and 4,4′-bpy has been reported [27]. The (benzotriazol-1-yloxy)-acetic acid is a good semi-rigid ligand for building supramolecular systems and it has abundant coordination modes due to the introduction of rich nitrogen heterocyclic ligands and carboxylate group. Meanwhile, due to the presence of abundant donor and accept atoms, it can form large number of hydrogen bonds and can be involved in π···π interactions, which can enable the construction of supramolecular structures. Therefore, we can speculate that the combination of 1-hydroxybenzotriazole and carboxylate groups in a ligand may lead to more interesting coordination structures. In order to further explore the (benzotriazol-1-yloxy)-acetic acid ligand on the formation of complexes, we selected (benzotriazol-1-yloxy)-acetic acid as the ligand to react with Zn(Ac)2 or Ni(Ac)2 and obtained two new complexes, namely [Zn(L)2(H2O)2]n, (1), and [Ni(L)2(H2O)2]n (2). The elemental analysis, IR spectra and PXRD patterns of both complexes have been determined and are reported here. In addition, the luminescent property of 1 and UV–Visible absorbance of 2 were also investigated and discussed. DFT calculations of the structures, stabilities, orbital energies, and composition characteristics of some frontier molecular orbitals of the two complexes were performed by means of Gaussian 09W package and taking B3LYP/lanl2dz basis set.
2 Experimental Section
2.1 Materials and Physical Measurements
All reagents and solvents used in the synthesis were purchased without further purification. HL ligand was prepared according to the literature of Huang et al. [27]. Elemental analyses for C, H and N were performed on a Perkin Elmer 240C elemental analyzer. FT-IR spectra were recorded on a Nicolet Impact 6700 spectrometer using KBr pellets in the range of 4000–400 cm− 1. Solid-state luminescent spectra were recorded with a SHIMAZU VF-320 X-ray fluorescence spectrophotometer at room temperature. Powder X-ray diffraction (PXRD) measurements were conducted on a Bruker D8 Advance X-ray diffractometer using Cu-Kα radiation (λ = 1.5406 Å), in which the X-ray tube was operated at 40 kV and 40 mA. Luminescent spectra were recorded with a Fluorlog-3 fluorescence spectrophotometer at room temperature.
2.2 Synthesis of [Zn(L)2(H2O)2]n (1)
A methanol solution (5 ml) of Zn(Ac)·2H2O (22.0 mg, 0.10 mmol) was added dropwise to a methanol solution (10 ml) of HL (38.6 mg, 0.20 mmol) and Et3N (0.05 ml). The reaction mixture was stirred at room temperature for 1 h to give a clear solution. Colorless crystals suitable for X-ray analysis were obtained through slow evaporation of the solution after 2 weeks (yield 45%, based on Zn). Anal. Calcd. (%) for C16H16ZnN6O8: C, 39.56; H, 3.32; N, 17.30. Found (%): C, 39.52; H, 3.38; N, 17.36. IR (KBr, ν/cm− 1): 3282(w), 1615(vs), 1420(s), 1319(m), 1276(w), 1160(w), 1108(m), 982(m), 753(m), 683(m), 569(w), 471(w).
2.3 Synthesis of [Ni(L)2(H2O)2]n (2)
The synthesis of complex 2 was similar to that of 1 but used Ni(Ac)2·4H2O (24.9 mg, 0.10 mmol) instead of Zn(Ac)·2H2O. Green block-shaped crystals were obtained (yield 39%, based on Ni). Anal. Calcd. (%) for C16H16NiN6O8: C, 40.12; H, 3.37; N, 17.54. Found (%): C, 40.07; H, 3.42; N, 17.36. IR (KBr, ν/cm− 1): 3272(w), 1614(vs), 1422(s), 1319(m), 1245(w), 1160(w), 1110(m), 980(m), 755(m), 690(m), 570(w), 472(w).
2.4 Crystal Structure Determination and Refinement
The crystal data of complexes 1 and 2 were collected on a Bruker Smart APEX II CCD diffractometer equipped with graphite monochromated Mo-Kα radiation (λ = 0.71073 Å) at 298(2) K. Multi-scan absorption corrections were applied using the SADABS program [28]. All structures were solved by direct method of SHELXS and refined by full-matrix least-squares fitting on F2 using SHELXL-2014 [29]. All non-hydrogen atoms were refined anisotropically. The organic hydrogen atoms were placed in calculated positions with fixed isotropic thermal parameters using a riding model. The water hydrogen atoms were located from a difference Fourier map and restrained with O–H = 0.850 Å. CCDC numbers 1834967 and 1834968 for complexes 1 and 2, respectively, contain the supplementary crystallographic data for this paper. Details of the crystal data and structure refinement for the two complexes are summarized in Table 1. Selected bond distances and angles are given in Table 2. The details of the hydrogen bonds are listed in Table 3.
2.5 Density Functional Theory (DFT) Calculations
All the theoretical calculations were carried out with the Gaussian 09 program [30] using the default convergence criteria. Minimum-energy structure of free HL ligand was calculated and confirmed through a frequency calculation. The full geometry optimizations of complexes 1 and 2 were performed starting from the X-ray crystal structure without any constraints on symmetry at density functional theory (DFT) level using the hybrid B3LYP functional [31, 32]. The calculations were performed by using the LANL2DZ basis set for metal atoms and the standard 6-31G* for C, H, O and N atoms.
3 Results and Discussion
3.1 Crystal Structure Description
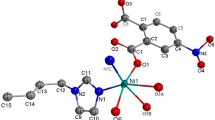
Single-crystal structure analysis reveals that complexes 1 and 2 are isostructural with different central metal ions, and crystallize in monoclinic crystal system P21/c with only slight differences in their bond lengths and angles. Hence, only the structure of complex 1 is described in detail here. The asymmetric unit of 1 contains one Zn(II) cation (located on a twofold axis of symmetry), one L ligand and one coordinated water molecule. As depicted in Fig. 1a, the Zn(II) center is six-coordinated by four oxygen atoms including two symmetry-related carboxylic oxygen atoms (O1i and O1iii; symmetry codes: (i) x, y, z − 1; (iii) − x + 1, − y + 1, − z + 1) and two symmetry-related water oxygen atoms (O1W and O1Wii; symmetry code: (ii) − x + 1, − y + 1, − z) and two symmetry-related nitrogen atoms (N1 and N1ii) at the axial position and at the equatorial positions. This gives an almost ideal {ZnN2O4} octahedral coordination geometry. The Zn–N and Zn–O bond lengths fall in the normal range of 2.2200(17) and 2.0700(13)-2.1096(14) Å, respectively, which are in the normal ranges.

The molecular structures of complex 1 with displacement ellipsoids drawn at the 40% probability level. Symmetry codes: (i) x, y, z − 1; (ii) –x + 1, − y + 1, − z; (iii) –x + 1, − y + 1, − z + 1. All H atoms have been omitted for clarity
In 1, neighboring Zn(II) cations are bridged by symmetrical L ligands to form an infinite one-dimensional linear chain with the Zn⋯Zn distance of 9.464 Å and Zn–Zn–Zn angle of 180° along the c axis (Fig. 2). In this 1D chain structure, each [Zn2(L)2] sixteen-membered metallamacrocycle is formed by two Zn(II) cations and two L ligands. Then, such chains are further linked by strong O1W–H1WA⋯O2iv (symmetry code: (iv) x, – y + 1/2, z − 1/2) hydrogen bonds (see Table 2 for full details), shaped by coordinated water molecules O1W and carboxylic oxygen O2, with O1W acting as donors and O2 as acceptors, to form a two-dimensional network parallel to the bc plane (Fig. 3). It is noted the adjacent chains in the two-dimensional network are arranged in an ⋯ABAB⋯ type. It is also noticed the strong intermolecular hydrogen bonds O1W–H1WB⋯O2i shaped by coordinated water molecules O1W and carboxylic oxygen O2 also exist and further stabilize the crystal structure. The hydrogen-bond details for 2 are given in Table 3.

A view of the one-dimensional structure of complex 1, along the c axis. All H atoms have been omitted for clarity

A view of the two-dimensional structure of complex 1, parallel to the bc plane linked by hydrogen bonds (purple dashed lines). Only H atoms involved in the interactions are shown. (Color figure online)
In the structure of 1, besides the hydrogen bonding interactions, offset face-to-face π⋯π interactions also present between the aromatic rings of neighbouring two-dimensional networks and play an important role in the crystal packing. By such π⋯π stacking interactions [Cg1⋯Cg1v = 3.633(5) Å. Cg1 is the centroid of the C1–C6 ring; symmetry code: (v) – x, – y + 1, – z], the neighboring two-dimensional networks are held together in a parallel ⋯AAAA⋯ fashion to generate a three-dimensional extended supramolecular framework in the solid state (Fig. 4).

a A view of the three-dimensional network of complex 1, viewed along the c axis. Only H atoms involved in the interactions are shown. Adjacent layers are shown in different colours. b The π⋯π interaction (green dashed line) between neighbouring two-dimensional sheets. (Color figure online)
In 2, offset face-to-face π⋯π interactions between the adjacent two-dimensional networks of [Ni(L)2(H2O)2] units [Cg1⋯Cg1v = 3.612(2) Å. Cg1 is the centroid of the C1–C6 ring; symmetry code: (v) – x + 1, – y, – z] also lead to the formation of the three-dimensional supramolecular structure.
3.2 Powder X-Ray Diffraction
Powder X-ray diffractions (PXRD) experiments were carried out for complexes 1 and 2 at room temperature to characterize their purities. As illustrated in Fig. 5a, b, the main peak positions of the simulated and experimental patterns match well with each other, demonstrating that the bulk synthesized materials and the measured single crystals are the same. The differences in intensity may be owed to the preferred orientation of the crystal samples.

The measured and simulated PXRD patterns of a complex 1 and b complex 2
3.3 Luminescent Properties of 1
The coordination polymers with d10 metal centers often exhibit luminescent properties and have various applications in photochemistry, light-emitting diodes and chemical sensor [33,34,35]. Therefore, the luminescent property of complex 1 in the solid state was investigated at room temperature. As shown in Fig. 6a, upon excitation at 260 nm, the intense broad emission band at 429 nm was observed. Moreover, at the same excitation wavelength, the free HL ligand presents a weaker photoluminescence emission at 434 nm, which can be probably attributed to the π–π* transitions. Obviously, the strong emission of 1 comes from the charge transfer from HL to metal and π–π* transition of coordinated HL. Compared to the free ligand HL, the emission of 1 was blue-shifted about 5 nm. In addition, the emission decay lifetime of 1 was monitored and the curve can be fitted with a three-exponential decay function: τ1 = 4.300 ns (24.00%), τ2 = 14.089 ns (2.94%), and τ3 = 1.023 ns (73.06%) (χ2 = 1.128, Fig. 6b).

a Photoluminescent spectra of free HL ligand and complex 1 at room temperature. b The fitted decay curve monitored at 429 nm for complex 1 in the solid state at room temperature. Blank circles: experimental data; Solid line: fitted by Fit = A + B1 × exp(– t/τ1) + B2 × exp(– t/τ2) + B3 × exp(– t/τ3)
3.4 UV–Visible Spectra of 2
In the previous literature, some coordination polymers have been reported to be potential semiconductors [36,37,38]. Inspired by these documents, the solid-state UV–Vis absorbance spectra of HL ligand and complex 2 are measured and displayed in Fig. 7a. The absorption peaks at 257 nm (HL) and 271 (2) can be ascribed to intraligand charge transfer transitions. In addition, two additional peaks are also observed for 2. The second small peak at 384 nm [3T1g(p) → 3A2g], and the third sites in 646 nm [3T1g(F) → 3A2g], which are typical for Ni(II) complexes with octahedral coordination geometry [11, 39, 40]. To investigate the conductivity potentials of complex 2, the diffuse reflectance data is transformed into Kubelka–Munk function [41], F = (1 − R)2/2R, to obtain its band gap (Eg), where R is the reflectance at a given energy. The F against E plots are shown in Fig. 7b, and Eg assessed from the linear portion of the absorption edge was estimated to be 2.60 eV for complex 2. The band gap value suggests that complex 2 possess the potential application used as semiconductive material.

a Solid state UV–Vis spectra of free HL ligand and complex 2 at room temperature. b Plot of Kubelka–Munk as a function of energy of the complex 2
3.5 DFT Study
According to the frontier molecular orbital theory, the frontier molecular orbitals play an important role in the electric and optical properties as well as in UV–Vis spectra and chemical reactions [42,43,44]. The single point energies and frontier molecular orbitals of complexes 1 and 2 together with free HL ligand were calculated. Figure 8 shows the isodensity surfaces of HOMO (the highest occupied molecular orbital) and LUMO (the lowest unoccupied molecular orbital) of the two complexes studied herein in comparison to free HL ligand. In free HL ligand, the HOMO electron-density distribution is mainly located at the benzotriazol ring (except N2 atom) and ethereal oxygen atom, while the LUMO electron-density distribution is mainly located at the benzotriazol ring. In contrast, in 1 and 2, the HOMO electron densities are strikingly similar and located at carboxylate ions and adjacent carbon atoms of two L ligands. But the LUMO electron-density distributions of 1 and 2 are big different. The LUMO electron-density distribution of 1 is mainly located at the benzotriazol ring and ethereal oxygen atom of two L ligands, while the LUMO electron-density distribution of 2 is mainly located at the benzotriazol ring and ethereal oxygen atom of two L ligands, carboxylate ions and adjacent carbon atoms of two L ligands, and Ni metal atom. For HL, the calculated energies of the HOMO (EHOMO) and LUMO (ELUMO) are − 6.594 and − 1.383 eV, respectively. As a result, the ΔE (LUMO–HOMO) gap in free HL is approximately 5.211 eV. For 1, the EHOMO is − 1.360 eV and ELUMO is 1.954 eV, with the ΔE (LUMO–HOMO) gap is 3.314 eV. For 2, the EHOMO is − 1.387 eV and ELUMO is 1.541 eV, with the ΔE (LUMO–HOMO) gap is 2.923 eV. The HOMO–LUMO gaps are significantly smaller for the complexes 1 and 2 than for the free HL ligand. But, these energy gaps are both in agreement with the value of 2–3 eV often encountered for stable transition metal complexes [45,46,47].

Frontier molecular orbitals and their energies for HL, complex 1 and complex 2
4 Conclusions
In conclusion, two new zinc(II) and nickel(II) complexes derived from a mixed N- and O-donor ligand have been synthesized and characterized by single-crystal X-ray diffraction, as well as by elemental analysis, IR spectroscopy, and UV–Vis and fluorescence spectroscopies. O–H⋯O hydrogen bonds and π⋯π stacking interactions play an important role in extending and stabilizing the three-dimensional supramolecular framework. DFT calculations show the show the two complexes have good stabilities.
References
M. Zhao, J. Su, J. Zhang, J.Y. Wu, Y.P. Tian, Acta Crystallogr. C 71, 799–803 (2015)
D. Zhao, D.J. Timmons, D. Yuan, H.C. Zhou, Acc. Chem. Res. 44, 123–133 (2011)
L.R. Mingabudinova, V.V. Vinogradov, V.A. Milichko, E. HeyHawkins, A.V. Vinogradov, Chem. Soc. Rev. 45, 5408–5431 (2016)
O.R. Evans, W. Lin, Acc. Chem. Res. 35, 511–522 (2002)
J.A. Sheikh, A. Clearfield, Inorg. Chem. 55, 8254–8256 (2016)
J.M. Frost, K.L.M. Harriman, M. Murugesu, Chem. Sci. 7, 2470–2491 (2016)
Z.G. Gu, C.H. Zhan, J. Zhang, X.H. Bu, Chem. Soc. Rev. 45, 3122–3144 (2016)
S.S. Zhao, J. Yang, Y.Y. Liu, J.F. Ma, Inorg. Chem. 55, 2261–2273 (2016)
A. Douvali, A.C. Tsipis, S.V. Eliseeva, S. Petoud, G.S. Papaefstathiou, C.D. Malliakas, I. Papadas, G.S. Armatas, I. Margiolaki, M.G. Kanatzidis, T. Lazarides, M.J. Manos, Angew. Chem. Int. Ed. 54, 1651–1656 (2015)
C.J. Doonan, C.J. Sumby, CrystEngComm 19, 4044–4048 (2017)
I. Nath, J. Chakraborty, F. Verpoort, Chem. Soc. Rev. 45, 4127–4170 (2016)
L. Radhakrishnan, J. Reboul, S. Furukawa, P. Srinivasu, S. Kitagawa, Y. Yamauchi, Chem. Mater. 23, 1225–1231 (2011)
M. Hu, J. Reboul, S. Furukawa, L. Radhakrishnan, Y. Zhang, P. Srinivasu, H. Iwai, H. Wang, Y. Nemoto, N. Suzuki, S. Kitagawa, Y. Yamauchi, Chem. Commun. 47, 8124–8126 (2011)
M.P. Suh, H.J. Park, T.K. Prasad, D.W. Lim, Chem. Rev. 112, 782–835 (2012)
W. Chaikittisilp, N.L. Torad, C. Li, M. Imura, N. Suzuki, S. Ishihara, K. Ariga, Y. Yamauchi, Chem. Eur. J. 20, 4217–4221 (2014)
R.R. Salunkhe, C. Young, J. Tang, T. Takei, Y. Ide, N. Kobayashi, Y. Yamauchi, Chem. Commun. 52, 4764–4767 (2016)
J. Tang, Y. Yamauchi, Nat. Chem. 8, 638–639 (2016)
W. Zhang, X. Jiang, Y. Zhao, A. Carné-Sánchez, V. Malgras, J. Kim, J.H. Kim, S. Wang, J. Liu, J.S. Jiang, Y. Yamauchi, M. Hu, Chem. Sci. 8, 3538–3546 (2017)
R.R. Salunkhe, Y.V. Kaneti, Y. Yamauchi, ACS Nano 11, 5293–5308 (2017)
M. Chen, H. Zhao, C.S. Liu, X. Wang, H.Z. Shi, M. Du, Chem. Commun. 51, 6014–6017 (2015)
C.E. Housecroft, Dalton Trans. 43, 6594–6604 (2014)
M. Du, C.P. Li, C.S. Liu, S.M. Fang, Coord. Chem. Rev. 257, 1282–1305 (2013)
L.S. Long, CrystEngComm 12, 354–1365 (2010)
H.Y. Lin, J. Luan, X.L. Wang, J.W. Zhang, G.C. Liu, A.X. Tian, RSC Adv. 4, 62430–62445 (2014)
R. Haldara, T.K. Maji, CrystEngComm 15, 9276–9295 (2013)
A.D. Burrows, CrystEngComm 13, 3623–3642 (2011)
D. Huang, H. Zhang, L. Sheng, X. Jiang, Z. Liu, H. Xu, Chin. J. Inorg. Chem. 30, 1641–1646 (2014)
Bruker, APEX2, SAINT and SADABS (Bruker AXS Inc, Madison, 2008)
G.M. Sheldrick, Acta Crystallogr. C 7, 3–8 (2015)
M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, J.A. Montgomery Jr., T. Vreven, K.N. Kudin, J.C. Burant, J.M. Millam, S.S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G.A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J.E. Knox, H.P. Hratchian, J.B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R.E. Stratmann, O. Yazyev, A.J. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, P.Y. Ayala, K. Morokuma, G.A. Voth, P. Salvador, J.J. Dannenberg, V.G. Zakrzewski, S. Dapprich, A.D. Daniels, M.C. Strain, O. Farkas, D.K. Malick, A.D. Rabuck, K. Raghavachari, J.B. Foresman, J.V. Ortiz, Q. Cui, A.G. Baboul, S. Clifford, J. Cioslowski, B.B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R.L. Martin, D.J. Fox, T. Keith, M.A. Al-Laham, C.Y. Peng, A. Nanayakkara, M. Challacombe, P.M.W. Gill, B. Johnson, W. Chen, M.W. Wong, C. Gonzalez, J.A. Pople (2009) Gaussian, Inc., Gaussian 09, Revision A.1, Wallingford, CT
A.D. Becke, J. Chem. Phys. 98, 5648–5652 (1993)
C. Lee, W. Yang, R.G. Parr, Phys. Rev. B 37, 785–789 (1988)
Z.Q. Shi, Y.Z. Li, Z.J. Guo, H.G. Zheng, Cryst. Growth Des. 13, 3078–3086 (2013)
Z.Q. Shi, Z.J. Guo, H.G. Zheng, Dalton Trans. 43, 13250–13258 (2013)
N.N. Ji, Z.Q. Shi, H.L. Hu, H.G. Zheng, Dalton Trans. 47, 7222–7228 (2018)
J. Guo, J.F. Ma, B. Li, W.Q. Kang, J. Yang, Cryst. Growth Des. 11, 3609–3621 (2011)
J. Guo, J.F. Ma, J.J. Li, J. Yang, S.X. Xing, Cryst. Growth Des. 12, 6074–6082 (2012)
W. Meng, Z. Xu, J. Ding, D. Wu, X. Han, H. Hou, Y. Fan, Cryst. Growth Des. 14, 730–738 (2014)
L. Qin, J.S. Hu, Y.Z. Li, H.G. Zheng, Cryst. Growth Des. 12, 403–413 (2012)
D. Sarma, K.V. Ramanujachary, S.E. Lofland, T. Magdaleno, S. Natarajan, Inorg. Chem. 48, 11660–11676 (2009)
W.W. Wendlandt, H.G. Hecht, In Reflectance Spectroscopy (Interscience Publishers, New York, 1966)
I. Fleming, Frontier Orbitals and Organic Chemical Reactions (Wiley, London, 1976)
B.Y. Liu, Z. Liu, G.C. Han, Y.H. Li, J. Mol. Struct. 975(1–3), 194–199 (2010)
G. Şükriye, Ö Namık, B.D. Tülay, Ü Bahri, D. Muharrem, A. Ömer, Polyhedron 29(12), 2393–2403 (2010)
P.J. Blower, J.R. Dilworth, R.I. Maurer, G.D. Mullen, C.A. Reynolds, Y.F. Zheng, J. Inorg. Biochem. 85(1), 15–22 (2001)
N. Süleymanoğlu, R. Ustabaş, Y.B. Alpaslan, U. Çoruh, S. Karakuş, S. Rollas, Struct. Chem. 21(2), 59–65 (2010)
Z.Q. Shi, N.N. Ji, R.G. Zhao, J.K. Li, Z.F. Li, Struct. Chem. 22(2), 225–233 (2011)
Acknowledgements
This work was supported by the Natural Science Foundation of Shandong Province of China (No. ZR2016BM14), Tai’an science and technology project (No. 2016GX1046), and the Postgraduate Foundation of Taishan University (No. Y2015-1-011).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ji, NN., Shi, ZQ. & Hu, HL. Two New Isomorphous Zinc(II) and Nickel(II) Supramolecular Coordination Polymers Based on the (benzotriazol-1-yloxy)-Acetic Acid Ligand: Syntheses, Structures, Properties, and DFT Analyses. J Inorg Organomet Polym 28, 2653–2661 (2018). https://doi.org/10.1007/s10904-018-0910-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10904-018-0910-z