
Abstract
Employing tetracarboxylate and imidazole mixed ligands to react with different transition meat salts afford two new 3D coordination polymers, {[Zn2(BPTC)(BBI)2]·(H2O)3·DMSO}n (1) and {[Cd2(BPTC)(BBI)2]·H2O}n (2) (H4BPTC = biphenyl-3,3′,4,4′-tetracarboxylic acid, BBI = 1,1′-(1,4-butanediyl)bis(imidazole)). Both these two coordination polymers are 4-connected network topologies. Compound 1 features a unprecedented 3-nodal (4,4,4)-connected network topology with the point symbol {64·82}, and 2 displays a (4,4)-connected binodal network bearing new topology with the point symbol of {72·84}2{73·83}. Additionally, thermal stability and photoluminescence properties of 1 and 2 were investigated.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Coordination polymers (CPs) have attracted considerable attention because of their potential applications and their intriguing topologies [1,2,3,4,5,6,7,8,9,10]. Up to now, although a large number of CPs have been reported, the construction of novel architectures still remain a challenge [11,12,13]. Ingenious designs and selection of organic ligands and the proper choice of metal ions are the main keys to achieve the target CPs [11]. The employment of mixed multidentate ligands has been demonstrated to be a facile strategy for the construction of novel architectures because that combining different ligands in a complex offers greater tunability of structural frameworks than using single ligands, and the different ligands have variable coordination modes and conformations [14,15,16,17,18,19,20]. On the other hand, the 4-connected topologies are concerned and very common in open-framework materials [21,22,23,24,25,26,27]. And numerous 4-connected nets such as dia, nbo, cds, pts, sra, sod, qtz,crb, gis, lon, irl, pcb, pcl, neb, unh, cag, gsi, mmt, zni, lvt, etc, have been explored [21,22,23]. Consequently, the assembly of 4-connected nets with new topologies will become increasingly difficult. In this study, we employed biphenyl-3,3′,4,4′-tetracarboxylic acid (H4BPTC) and 1,1′-(1,4-butanediyl)bis(imidazole) (BBI) as the mixed ligands to successfully construct two new 3D CPs, namely {[Zn2(BPTC)(BBI)2]·(H2O)3·DMSO}n (1) and {[Cd2(BPTC)(BBI)2]·H2O}n (2), which display two types of unprecedented network topologies, and their thermal stability as well as photoluminescence properties were investigated.
2 Experimental
2.1 Materials and Physical Measurements
All reagents and H4BPTC were obtained commercially and used without further purification. The BBI ligand was prepared according to the literature [28]. Elemental analysis for C, H, and N were performed on a Perkin-Elmer 240 elemental analyzer. The FT-IR spectra were recorded from KBr pellets in the range from 4000 to 400 cm−1 on a Nicolet-5700 FTIR spectrometer. Thermogravimetric analyses (TGA) were collected on a TA SDT-Q600 thermogravimetric analyzer from room temperature with a heating rate of 10 °C min−1 under nitrogen. Powder X-ray diffraction (PXRD) were measured on a Puxi XD-3 diffractometer (Cu-Kα, λ = 1.5406 Å) at room temperature. Fluorescence spectra for the solid samples were measured on a Hitachi F–7000 fluorescence spectrophotometer at room temperature.
2.2 Syntheses of Compounds 1 and 2
2.2.1 Synthesis of {[Zn2(BPTC)(BBI)2]·(H2O)3·DMSO}n (1)
Zn(NO3)2·6H2O (0.0178 g, 0.06 mmol), Biphenyl-3,3′,4,4′-tetracarboxylic acid (0.010 g, 0.03 mmol), BBI (0.0057 g, 0.03 mmol), N,N-Dimethylformamide (0.75 mL), DMSO (1.5 mL), and HNO3 (0.25 mL, 1.31 M in H2O) added respectively to a 25-mL vial, which was sealed and heated to 85 °C for 72 h and then cooled to room temperature. Colorless block crystals were collected. IR (KBr, cm−1): 3449(s), 3116(s), 3033(w), 2943(m), 1614(s), 1343(s), 1295(w), 1246(m), 1087(s), 1031(m), 949(s), 830(s), 789(m), 761(m), 657(s), 588(w), 497(w).
2.2.2 Synthesis of {[Cd2(BPTC)(bbi)2]·H2O}n (2)
Cd(NO3)2·4H2O (0.0344 g, 0.11 mmol), Biphenyl-3,3′,4,4′-tetracarboxylic acid (0.0165 g, 0.05 mmol), BBI (0.0297 g, 0.16 mmol), N,N-Dimethylformamide (2.45 mL), H2O (0.5 mL), and HNO3 (0.30 mL, 2.40 M in DMF) added respectively to a 25-mL vial, which was sealed and heated to 85 °C for 24 h and then cooled to room temperature. Colorless block crystals were collected. IR (KBr, cm−1): 3428(s), 3122(s), 3026(w), 2936(m), 1641(m), 1558(s), 1391(s), 1246(m), 1156(w), 1101(s), 941(s), 837(s), 789(m), 755(m), 664(s), 582(w), 449(s).
2.3 Crystal Structure Determination
Single-crystal X-ray diffraction Data collection was performed on a Bruker SMART CCD diffractometer equipped with a graphite monochromated Mo Kα radiation (λ = 0.71073 Å). All calculations were carried out on a computer with the use of SHELXL-97 programs [29]. The structures were solved by direct methods and refinement on F2 using the full-matrix least-squares methods. Detailed crystallographic data and structure refinement parameters of compounds 1 and 2 are given in Table 1.
3 Results and Discussion
3.1 Description and Comparison of Crystal Structures
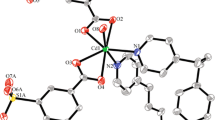
Colorless crystals of {[Zn2(BPTC)(BBI)2]·(H2O)3·DMSO}n (1) were prepared via solvothermal reaction of Zn(NO3)2·6H2O with biphenyl-3,3′,4,4′-tetracarboxylic acid and 1,4-bis(imidazol-1-ylmethyl)-benzene (Scheme 1). Single-crystal X-ray structural analysis reveals that 1 crystallizes in orthorhombic, Pbca space group. Each asymmetric unit contains two crystallographically independent Zn atoms, one BPTC ligand, two BBI ligands, one DMSO, and three H2O molecules. As shown in Fig. 1a, Zn atoms are all tetrahedrally coordinated by two N atoms from BBI ligands and two O atoms from BPTC ligands. Each BPTC adopts the same cis conformation, and its four carboxylates coordinated to four distinct Zn atoms in a monodentate mode. Zn1 and Zn2 centers link with each other through BPTC ligands to form a herringbone layer along the ac plane (Fig. 1b). The herringbone layers are supported by the BBI pillars in two different modes via the N-Zn coordination bonds into a 3D pillared-layer structure with 1D channels (Fig. 1c). These channels are actually filled by the DMSO and H2O molecules. When these extraframework species are not considered, the total accessible volume within the crystal is estimated as 19.2% as calculated by PLATON [30]. The topological method was applied to explore the nature of this fascinating structure. Zn1 and Zn2 atoms can be considered as two kinds of 4-connected nodes, the cis BPTC ligands serve as 4-connected T-shaped nodes, and the BBI ligands are simplified to be linear connectors. As a result, the structure of 1 shows a novel 3-nodal (4,4,4)-connected network topology with the point symbol {64·82}, as shown in Fig. 1d.

The ligands H4BPTC and BBI

a Environments of metal centers and ligands in 1; b view of the 2D herringbone sheet of 1; c view of the 3D pillar-layer framework of 1 (BBI with different conformations are marked as green and blue. The pink and yellow balls representation of H2O and DMSO, respectively); d view of the topological representation of the 3-nodal (4,4,4)-connected network of 1. All the hydrogen atoms are omitted for clarity
Under similar reaction conditions of 1, when using cadmium salt instead zinc salt, large block colorless crystals of 2 are obtained. Examination of the structure of 2 suggests that it shows an alternative unprecedented structural topology. 2 crystallizes in P21/c space group. The asymmetric unit contains two crystallographically nonequivalent Cd atoms (Cd1 and Cd2), one BPTC, two BBI and one lattice H2O. As illustrated in (Fig. 2a), Cd1 is six-coordinated with distorted octahedral environment by two N atoms from two BBI ligands and four O atoms from two different BPTC ligands, and Cd2 is surrounded by two N atoms from two different BBI ligands and three O atoms from two individual BPTC ligands. Each BPTC ligand adopts a µ4-bridging mode using its four deprotonated carboxylate groups to link four Cd centers. As have been observed in Fig. 2b, Cd1 and Cd2 are bridged respectively by 3,4- and 3′,4′-carboxylate groups of BPTC to give a herringbone layer. The herringbone layers are supported by the BBI pillars in two different modes via the N-Zn coordination bonds into a 3D pillared-layer structure with 1D channels (Fig. 2c), and the channels are filled by H2O. The porosity of 2 calculated by PLATON is 5.0% of the unit cell (with water molecules removed). It is worth mentioning that these BBI ligands show three connected modes, where two BBI ligands act as pillars between two adjacent layers, another one BBI act as ancillary terminal ligand to coordinate to Cd centers of metal-carboxylate layers. In a view of topology, Cd1 and Cd2 atoms can be considered as two kinds of 4-connected nodes, the trans BPTC ligands serve as 4-connected nodes, and the BBI ligands are simplified to be linear connectors. On the basis of simplification, 2 possesses a novel 3D binodal (4,4)-connected network topology with the point symbol of {72·84}2{73·83} (Fig. 2d). Although numerous 4-connected networks have been reported, to our best knowledge, the two kinds of topology defined by compounds 1 and 2 have not been observed [21,22,23,24,25,26,27, 31,32,33].

a Environments of metal centers and ligands in 2; b view of the 2D herringbone sheet of 2; c view of the 3D pillar-layer framework of 2 (BBI with different conformations are marked as green, blue, and gray. The pink balls representation of H2O); d view of the topological representation of the 3-nodal (4,4)-connected net work of 2. All the hydrogen atoms are omitted for clarity
The comparison of 1 and 2 reveals that they contain the same herringbone metal–carboxylate layer, but where BPTC ligands show different conformations, and the tetracarboxylate groups of BPTC present different coordination modes with metal centers. Notably, BPTC ligands in 1 exhibit rare cis conformation [34, 35]. Additionally, BBI ligands show difference connected modes, all BBI ligands in 1 only act as pillars to bridge the neighboring metal–carboxylate layers; However, different from 1, BBI ligands in 2 not only act as pillars, but also act as ancillary terminal ligands to consolidate the metal–carboxylate layers. On the other hand, 1 and 2 were prepared under similar conditions only by varying metal salts, but they possess different structural topologies. The results imply that the metal centers have a remarkable impact on the structure of the compounds due to their different coordination geometric requirements.
3.2 IR of Compounds 1 and 2
To find out more information about the organic moieties and their binding to the metal centers, the title compounds have been analyzed by FT-IR spectroscopy. The IR spectra of the two compounds are shown in Fig. 3. For 1, the broad band centered at 3441 cm−1 shows the presence of water molecules. The absorption bands in the region between 3101 and 2867 cm−1 can be assigned to stretching vibrations of the ν(C–H) and ν(N–H) bonds of the aromatic rings. In the low-frequency range of 955–582 cm−1, the characteristic peaks of the aromatic ring are observed. The Zn-oxygen core vibrations are weak and observed at 443 cm−1. The strong characteristic absorptions at 1620 and 1364 cm−1 can be assigned to the asymmetric stretching and symmetric stretching vibrations of the carboxylate groups, respectively. The separation (Δ) between νasym(COO) and νsym(COO) is at 256 cm−1, suggests a monodentate binding of the carboxylate groups to the metal ions [36]. For 2, its characteristic peaks are similar to them of 1 except for carboxylate groups’ characteristic peaks. The bands at about 3428 cm−1 can be assigned to the characteristic peaks of water molecules. The ν(C–H) and ν(N–H) characteristic peaks of the aromatic rings are in the region of 3130–2860 cm−1. The characteristic peaks of the aromatic ring appear in the 941–575 cm−1 region, and the peak about 450 cm−1 corresponds to the Cd-oxygen core vibrations. Compared with the characteristic absorptions of carboxylate groups, there is some difference between 1 and 2. In 2 the asymmetric stretching peaks locate at 1648 and 1550 cm−1, and the symmetric stretching peak is at 1406 cm−1. The Δ are at 242 and 144 cm−1, respectively, which is attributed to the co-existence of the monodentate and bridging modes of the carboxylate groups in 2 [36]. The IR spectra indicate that the BPTC4− carboxylate groups in 1 and 2 function in different coordination modes, being in good agreement with crystal structural analyses.

FT-IR spectra of compounds 1 and 2
3.3 TGA and PXRD of Compounds 1 and 2
Thermogravimetric analysis (TGA) was carried out under N2 atmosphere from room temperature to 800 °C, and results were showed in Fig. 4a. The TGA curve of 1 shows the first weight loss of 5.45% from 28 to 75 °C, which corresponds to the loss of three lattice water molecules (calculated 5.58%). A weight loss of 8.15% occurs between 185 and 256 °C, which corresponds to the loss of one DMSO molecule (calculated 8.05%), and decomposition of the organic ligands occurs above 295 °C. Finally, 1 was completely degraded into ZnO at 480 °C with total loss of 83.21% (calcd. 83.45%). For 2, the weight loss of 2.52% was observed between room temperature and 202 °C, which is attributed to the release of the water molecules, but it is slightly higher than the calculated value of 1.90%. The reason is that 2 adsorbed some water molecules on its crystal surface. The framework decomposed at 310 °C along with the release of the BPTC and BBI ligands, and then the complex was converted to a mixture containing CdO at 800 °C with the residual weight of 30.61%, which is not in accordance with the calculated value (27.05%). The reason is that the mixture contains C except for Cd and O as estimated by elemental analysis, and some similar examples have been reported [37, 38].

a TGA curves of compounds 1 and 2; b PXRD patterns of compounds 1 and 2
The powder X-ray diffraction (PXRD) patterns of compounds 1 and 2 were also investigated. The simulated and experimental PXRD patterns of 1 and 2 are shown in Fig. 4b indicating the phase purity of the products. The peak positions of simulated and experimental patterns are in good agreement with each other; the differences in intensity may be due to the preferred orientation of the powder samples [39].
3.4 Photoluminescence Properties of Compounds 1 and 2
The solid-state photoluminescence spectrum of compounds 1 and 2 were measured, and are compared with the free H4BPTC and BBI ligands (Fig. 5). The free H4BPTC and BBI ligands exhibit fluorescent emission peaks around 393 nm (ex = 351 nm) and 411 nm (ex = 346 nm), respectively, which are attributed to π*–n or π*–π (intraligand electron) transition. Upon excitation at 346 and 355 nm, compound 1 and 2 show blue fluorescent emission band around 397 and 423 nm, respectively. Comparing the emission peak of free H4BPTC and BBI ligands, the fluorescent emission of 1 and 2 can be tentatively assigned to intraligand transfer π*−π transitions of H4BPTC because similar peak also appear for the CPs built from H4BPTC [40, 41]. The red shift of the emission in 1 and 2 in comparison to H4BPTC ligand should be ascribed to the metal–ligand coordinative interactions [40,41,42].

Solid-state emission spectra for free H4BPTC, BBI, 1 and 2 at room temperature
4 Conclusions
Employing tetracarboxylate and imidazole mixed ligands, two new 3D CPs have been successfully obtained by varying metal salts. Compounds 1 and 2 contain the same herringbone metal–carboxylate layer, but they display two different unprecedented 4-connected network topologies. The results show that the metal centers have a remarkable impact on the structure of the compounds due to their different coordination geometric requirements. And 1 and 2 show fluorescence in the solid state, which result from intraligand transfer π*–π transitions of H4BPTC. This study demonstrates new types of structural topologies, which are essential for providing insight into topology motifs and complicated structures.
5 Supplementary Materials
CCDC 1042391 and 1562852 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
References
H. Furukawa, K.E. Cordova, M. O’Keeffe, O.M. Yaghi, Science. 341, 1230444 (2013)
C. Janiak, J.K. Vieth, New J. Chem. 34, 2366 (2010)
M.L. Hu, V. Safarifard, E. Doustkhah, S. Rostamnia, A. Morsali, N. Nouruzi, S. Beheshti, K. Akhbari, Microporous Mesoporous Mater. 256, 111 (2018)
S. Rostamnia, F. Mohsenzad, Mol. Catal. 445, 12 (2018)
S. Rostamnia, H. Alamgholiloo, M. Jafari, R. Rookhosh, A.R. Abbasi, Appl. Organometal. Chem. 30, 954 (2016)
S. Rostamnia, H. Alamgholiloo, X. Liu, J. Colloid Interface Sci. 469, 310 (2016)
S. Rostamnia, Z. Karimi, Inorg. Chim. Acta. 428, 133 (2015)
S. Rostamnia, H.C. Xin, Appl. Organometal. Chem. 28, 359 (2014)
S. Rostamnia, A. Morsali, RSC Adv. 4, 10514 (2014)
S. Rostamnia, A. Morsali, Inorg. Chim. Acta. 411, 113 (2014)
J.J. Perry, I.V.,J.A. Perman, M.J. Zaworotko, Chem. Soc. Rev. 38, 1400 (2009)
M. Li, D. Li, M. Ókeeffe, O.M. Yaghi, Chem. Rev. 114, 1343 (2014)
M. Alipour, O. Akintola, A. Buchholz, M. Mirzaei, H. Eshtiagh-Hosseini, H. Görls, W. Plass, Eur. J. Inorg. Chem. 5356 (2016)
L.J. Liu, K. Konstas, M.R. Hill, S.G. Telfer, J. Am. Chem. Soc. 135, 17731 (2013)
H. Zhang, C.H. Zhou, D. Liu, G.Y. Li, J. Inorg. Organomet. Polym. Mater. 27, 923 (2017)
Q.F. Zhang, A.J. Geng, H.N. Zhang, F.L. Hu, Z.H. Lu, D.Z. Sun, X.L. Wei, C.L. Ma, Chem. Eur. J. 20, 4885 (2014)
J.S. Hu, Y.J. Shang, X.Q. Yao, L. Qin, Y.Z. Li, Z.J. Guo, H.G. Zheng, Z.L. Xue, Cryst. Growth Des. 10, 2676 (2010)
F. Yu, B. Li, CrystEngComm. 13, 7025 (2011)
Y. Wang, Y. Qi, V.A. Blatov, J.M. Zheng, Q. Li, C. Zhang, Dalton Trans. 43, 15151 (2014)
G.L. Li, W.D. Yin, G.Z. Liu, L.F. Ma, L.Y. Wang, J. Solid State Chem. 220, 1 (2014)
D.S. Li, Y.P. Wu, J. Zhao, J. Zhang, J.Y. Lu, Coord. Chem. Rev. 261, 1 (2014)
R. Natarajan, G. Savitha, P. Dominiak, K. Wozniak, J.N. Moorthy, Angew. Chem. Int. Ed. 44, 2115 (2005)
I.A. Baburina, V.A. .Blatova, L. Carluccib, G. Cianib, D.M. Proserpiob, J. Solid State Chem. 178, 2452 (2005)
L.D. Luan, H.M. Zeng, Z.E. Lin, H.Q. Cai, H. Huang, Inorg. Chem. Commun. 78, 5 (2017)
L.F. Ma, Y.Y. Wang, J.Q. Liu, G.P. Yang, M. Du, L.Y. Wang, CrystEngComm. 11, 1800 (2009)
L.F. Ma, C.P. Li, L.Y. Wang, M. Du, Cryst. Growth Des. 11, 3309 (2011)
L. Qin, S.Y. Zeng, W.J. Zuo, Q.H. Liu, J. Li, G. Ni, Y.Q. Wang, M.D. Zhang, Polyhedron. 121, 231 (2017)
J.F. Ma, J. Yang, G.L. Zheng, L. Li, J.F. Liu, Inorg. Chem. 42, 7531 (2003)
G.M. Sheldrick, SHELXTL–97, Program for the Solution of Crystal Structures, (University of Göttingen, Germany, 1997)
A.L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr. 65, 148 (2009)
V.A. Blatov, A.P. Shevchenko, D.M. Proserpio, Cryst. Growth Des. 14, 3576 (2014)
O. Delgado-Friedrichs, M.D. Foster, M. ÓKeeffe, D.M. Proserpio, M.M.J. Treacy, O.M. Yaghi, J. Solid State Chem. 178, 2533 (2005)
L. Öhrström, Chem. Eur. J. 22, 13758 (2016)
S.J. Deng, N. Zhang, W.M. Xiao, C. Chen, Inorg. Chem. Commun. 12, 157 (2009)
G.D. Feng, L. Jiang, Q. Chen, D.S. Yang, M.J. Wang, Z.X. Li, X.L. Luo, Chin. J. Struct. Chem. 32, 45 (2013)
G.B. Deacon, R.J. Phillips, Coord. Chem. Rev. 33, 227 (1980)
S.M. Ding, Q.L. Dong, J.W. Hu, W.M. Xiao, X.H. Liu, L.Q. Liao, N. Zhang, Chem. Commun. 52, 9757 (2016)
R.N. Ma, C. Chen, B. Sun, X.T. Zhao, N. Zhang, Inorg. Chem. Commun. 14, 1532 (2011)
L.L. Zhang, J. Guo, Q.G. Meng, H.D. Pang, Z. Chen, D.F. Sun, Inorg. Chem. Commun. 26, 51 (2012)
S.J. Deng, W.T. Chao, F.Q. Huang, L. Gan, X.P. Duan, T.X. Liu, W.M. Xiao, N. Zhang, Z. Anorg. Allg. Chem. 640, 1551 (2014)
G.X. Liu, K. Zhu, H. Chen, R.Y. Huang, X.M. Ren, Z. Anorg. Allg. Chem. 635, 156 (2009)
F. Wang, X.H. Ke, J.B. Zhao, K.J. Deng, X.K. Leng, Z.F. Tian, L.L. Wen, D.F. Li, Dalton Trans. 40, 11856 (2011)
Acknowledgements
This work was supported by the National Natural Science Foundations of China (No. 21263014), the Natural Science Foundations of Jiangxi Province (Nos. 2011ZBAB203013 and 20122BAB203010), and the Foundation of Educational Department of Jiangxi Province (No. GJJ12103).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest
Electronic Supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, T., Ji, J., Deng, S. et al. Two 3D Coordination Polymers with New Topology Based on Mixed Ligands: Synthesis, Structure, and Photoluminescence Properties. J Inorg Organomet Polym 28, 1439–1445 (2018). https://doi.org/10.1007/s10904-018-0810-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10904-018-0810-2