
Abstract
In this study, we investigated a new series of naphthalimide based Schiff base compounds as potential DNA binding, antioxidant and antimicrobial agents. The structural characterization of synthesized compounds was carried out with the aid of elemental analysis and spectroscopic techniques (UV–vis., IR, 1H and 13C NMR). The DNA binding properties of target compounds against Ct-DNA (calf thymus) have been investigated in detail by numerous biophysical techniques (UV–vis, fluorescence, ethidium bromide displacement assay, Time resolved fluorescence, viscosity, cyclic voltammetry and circular dichorism) and the evidences have suggested that the test compounds could interact with DNA via intercalative binding. The extent of DNA binding (Kb) of these compounds follow the order of 3b (3.33 × 104 M−1) > 3a (2.25 × 104 M−1) > 3c (2 × 104 M−1), suggesting that compound 3b binds more strongly to Ct- DNA than the compounds 3a and 3c. Molecular docking results further support intercalative binding of test compounds with DNA. The binding energies of docked compounds (3a-3c) were found to be −8.20 to −8.69 kcal/ mol, suggesting greater binding affinity to Ct-DNA. The synthesized compounds displayed potential antimicrobial activities against Escherichia coli, Staphylococcus aureus, Klebsiella pneumoniae and Salmonella typhimurium. Compound 3c has emerged as most active against all the four tested bacterial strains with MIC value in the range of 0.031–0.062 mg/mL. In the mutagenicity studies, all the test compounds were found to be non-mutagenic both in the presence and absence of metabolic activation. Furthermore, the antioxidant activity experiments show that these compounds exhibited potential scavenging activities against DPPH and H2O2 radicals.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Naphthalimides are important targets for drug discovery because of their applications for bioactivities, such as antitumor, antibacterial, antimalarial, photobiological potency and DNA photocleaving activities [1–5]. Naphthalimides have been extensively studied as DNA intercalators and topo II inhibitors [6]. Moreover, a series of naphthalimides have been recently demonstrated as clathrin [7] and β-GlcNAcase inhibitors. Apart from their significance in the medicinal chemistry, naphthalimides have found many important applications as fluorescent dyes [8], sensors [9] and organic light emitting materials [10]. Owing to their planar aromatic ring and hydrophobicity naphthalimides could easily interact with a biological polymer (DNA, RNA, proteins, etc.) which constitutes the basis for biological activity [11]. Therefore, considerable effort has been increasingly given to the development of most potent naphthalimide derivatives in drug chemistry [12].
Over the past decades, search for chemotherapeutic anticancer drugs that can target specific DNA sequences has become an important theme in cancer chemotherapy. It was well documented that the effectiveness of anticancer drugs credited to their DNA-interacting capabilities [13]. Therefore, special attention has been devoted over the past few years in the studies of drug-DNA interactions. Numerous techniques have been employed to detect the binding mode between the small molecules and DNA, including fluorescence spectroscopy [14], ultraviolet–visible spectroscopy [15], nuclear magnetic resonance [16], electrochemistry [17], mass spectrometry [18], circular dichroism [19] and hydrodynamic measurements [20]. Currently, molecular docking study has been widely used in modern drug designing to investigate the binding mode between small molecule drug candidates and receptors [21].
Significant progress has been made over the past decades for the synthesis of naphthalimide derivatives as DNA intercalators (Fig. 1). Among these, amonafide (5-amino substituted naphthalimide) represent earliest and perhaps most extensively studied as DNA intercalator and exhibited pronounced antitumor activity against advanced breast cancer [22]. Anticancer activity of naphthalimides has been extensively studied against a number of different human tumor cell lines [23]. However, significant side effects and poor tumor selectivity related to these compounds has prompted researchers to search continuously for new antitumor agents. The cytotoxic efficiency of naphthalimides may be mainly attributed to the DNA intercalation. Therefore, the study of the interaction of naphthalimides with Ct-DNA is considered of great significance in the investigation of their potential of anticancer activities. Detection of genotoxicity for new chemicals has been attractive in order to find out their potential to cause damage to DNA, which in turn causes gene mutations [24]. Nowadays, in vitro and in vivo genotoxicity tests for new chemicals have been desirable to evade the threat of genotoxic exposure to mutagens and toxicity [25]. Ames test has been routinely used to evaluate the potential of new chemicals to cause gene mutations and chromosomal alterations. In view of the significance of naphthalimides for further therapeutic evolution, it is appropriate to assess the genotoxic activities of these compounds.

Structures of amonafide and N-acetylamonafide
The biological significance of Schiff base ligands in the medicinal chemistry has been known for several decades. Many studies have been investigated regarding the biological activities of Schiff bases, including their anticancer [26], antibacterial [27], antifungal [28] and antiviral activities [29]. Because of their good antimicrobial activity, Schiff base can be incorporated into naphthalimide pharmacophore to enhance the overall activity. Although, the pharmacological activities of naphthalimides have been paid more attention long before, the antimicrobial activities of naphthalimides linked to Schiff base have few been reported. As part of our on-going work, we herein described the antimicrobial activities of some novel N-substituted naphthalimide derivatives which have not been reported so far.
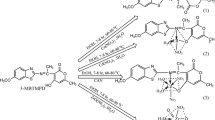
The general synthetic route we applied for the synthesis of the napthalimide analogues 3a-3c is summarized in scheme 1. The structural characterization of these compounds was carried out by IR, 1H NMR, 13C NMR and UV–vis. spectral analysis. The interaction ability of the target compounds with native Ct-DNA has been studied by means of UV-visible, fluorescence spectroscopy, circular dichroism, electrochemical and thermodynamic measurements.

The synthetic route of N-substituted naphthalimide derivatives
Experimental Section
Materials and Methods
All reagents were used as purchased from Sigma-Aldrich. The solvents were of spectroscopy grade and used without further treatment. Ct- DNA were purchased from SRL (India), Tris–HCl, were purchased from Merck. Double distilled water was used to prepare all buffer solutions. The reactions were monitored by thin layer chromatography using UV cabinet for visualization. Melting point was recorded using an electro-thermal melting point apparatus and were uncorrected. Elemental analysis was carried out on a Vario-Micro elemental analyzer. Electronic spectra were achieved with Perkin Elmer Lambda 40 UV-Visible spectrophotometer. IR spectra were recorded in the range of 4000–400 cm−1 on a Perkin Elmer Spectrum RXI IR instrument as KBr discs. 1H NMR spectra were recorded on Bruker DPX-300 NMR spectrometer operating at 300 MHz using DMSO-d6 as solvent. 13C NMR spectra were recorded on Agilent-NMR-vnmrs500 spectrometer. Fluorescence spectra were obtained on Jobin-Yvon Fluorolog 322 spectrofluorometer using 450 W xenon lamp and R928P PMT as the excitation source and detector, respectively. CD spectra were obtained on a Chirascan CD spectropolarimeter. Cyclic voltammetric measurements were accomplished on a DY2312 potentiostat.
Synthesis
Preparation of 2-amino-benzo[de]isoquinolin-1,3-dione (2)
2-Amino-benzo[de]isoquinolin-1,3-dione (2) has been synthesized according to reported procedure [30].
Yellow solid; Yield 74 %; m.p: 258 °C. UV λmax (nm): 333, 241. IR ʋmax(cm−1): 3312, 3238 ʋ(NH2); 1775, 1707, ʋ(C = O); 732 ʋ(Ar); 1H NMR (300 MHz, DMSO-d6) δ in ppm: 8.47 (d, 2H, J = 7.2 Hz, Ar-H); 8.42 (d, 2H, J = 8.4 Hz, Ar-H); 7.86 (t, 2H, J = 7.8 Hz, Ar-H); 5.77 (s, 2H, −NH2). 13C NMR (100 MHz, DMSO-d6) δ in ppm: 160.94; 134.94; 131.70; 131.22; 127.73; 126.43; 122.13; Anal. Calcd. for C21H16N2O4 (212.2): C, 67.92; H, 3.80; N, 13.20. Found: 67.95; H, 3.84; N, 13.17.
General procedure for synthesis of compounds (3a-3c)
To a solution of 2-amino-benzo[de]isoquinolin-1,3-dione (0.424 g, 2 mmol) in 50 mL of DMF were added appropriate aldehyde (2.2 mmol) in absolute EtOH and few drops of glacial acetic acid. The reaction mixture stirred upon heating for 6–8 h. Then it was poured onto ice, and the resulting yellow precipitate was filtered through a buchner funnel and washed with cold ethanol. Recrystallization with aqueous DMF provided pure compounds. Finally the compounds dried in vacuum desiccators over fused calcium chloride.
2-[(2-Hydroxy-3-methoxy)-benzylidene)amino]benzo[de]isoquinolin-1,3-dione (3a)
Yellow solid; Yield 65 %; m.p: 274 °C. UV λmax (nm): 339, 237. IR ʋmax (cm−1): 3253 ʋ(OH); 2933, 2862, ʋ(CH); 1782, 1730, ʋ(C = O); 1626 ʋ(C = N); 739 ʋ(Ar); 1H NMR (300 MHz, DMSO-d6) δ in ppm: 10.65 (br, 1H, −OH); 9.01 (s, 1H, −CH = N); 8.53 (t, 4H, J = 6.9 Hz, Ar-H); 7.92 (t, 2H, J = 6.9 Hz, Ar-H); 7.42 (d, 1H, J = 7.5 Hz, Ar-H); 7.22 (d, 1H, J = 6.9 Hz, Ar-H); 6.99 (t, 1H, J = 7.5 Hz, Ar-H); 3.87 (s, 3H, −OCH3); 13C NMR (125 MHz, CdCl3) δ in ppm: 160.93; 151.94; 151.12; 146.57; 134.43; 131.52; 131.49; 130.17; 127.31; 127.08; 126.72; 121.84; 114.29; 109.96; 56.16; Anal. Calcd. for C20H14N2O4 (346.34): C, 69.36; H, 4.07; N, 8.09. Found: C, 69.32; H, 4.11; N, 8.12.
2-[(4-Hydroxy-3-ethoxy)-benzylidene)amino]benzo[de]isoquinolin-1,3-dione (3b)
Brown solid; Yield 61 %; m.p: 256 °C. UV λmax (nm): 340, 226. IR ʋmax (cm−1): 3260 ʋ(OH); 2903, 2862, ʋ(CH); 1823, 1722, ʋ(C = O); 1640 ʋ(C = N); 736 ʋ(Ar); 1H NMR (300 MHz, DMSO-d6) δ in ppm: 10.05 (br, 1H, −OH); 9.75 (s, 1H, −CH = N); 8.54 (d, 2H, J = 8.1 Hz, Ar-H); 8.53 (d, 2H, J = 6.9 Hz, Ar-H); 7.93 (t, 2H, J = 7.5 Hz, Ar-H); 7.41 (dd, 1H, J = 6.3,1.8 Hz, Ar-H); 7.36 (s, 1H, − Ar-H); 6.97 (d, 1H, J = 8.1 Hz, Ar-H); 4.13 (q, 2H, J = 6.6 Hz, −OCH2); 1.38 (t, 3H, J = 6.9Hz,-CH3). 13C NMR (125 MHz, CdCl3) δ in ppm: 160.95; 151.82; 150.84; 146.43; 134.47; 131.57; 131.52; 129.82; 127.29; 127.02; 126.77; 121.86; 114.37; 109.61; 64.76; 14.67; Anal. Calcd. for C21H16N2O4 (360.36): C, 69.99; H, 4.48; N, 7.77. Found: 69.95; H, 4.53; N, 7.75.
2-[(3-Methoxy-4-methoxy)-benzylidene)amino]benzo[de]isoquinolin-1,3-dione (3c)
Yellow solid; Yield 68 %; m.p: 248 °C. UV λmax (nm): 337, 233. IR ʋmax (cm−1): 2932, 2862, ʋ(CH); 1789, 1730, ʋ(C = O); 1640 ʋ(C = N); 736 ʋ(Ar); 1H NMR (300 MHz, DMSO-d6) δ in ppm: 8.59 (s, 1H, −CH = N); 8.54 (d, 2H, J = 7.5 Hz, Ar-H); 8.50 (d, 2H, J = 8.1 Hz, Ar-H); 7.93 (t, 2H, J = 7.5 Hz, Ar-H); 7.59 (s, 1H, Ar-H); 7.46 (d, 1H, J = 8.1 Hz, Ar-H); 7.16 (d, 1H, J = 8.1 Hz, Ar-H); 3.87 and 3.85 (s, 3H, 2(OCH3)); 13C NMR (125 MHz, CdCl3) δ in ppm: 160.87; 154.47; 151.72; 149.61; 134.40; 131.64; 131.54; 130.13; 126.98; 126.80; 126.69; 121.86; 110.39; 108.96; 56.14; Anal. Calcd. for C21H16N2O4 (360.36): C, 69.99; H, 4.48; N, 7.77. Found: C, 69.94; H, 4.51; N, 7.73.
DNA Binding Studies
Absorption Titrations
Absorbance spectroscopy is an optical technique useful in examining the interaction between small molecules and nucleic acids. The relative binding of test compounds with Ct-DNA was performed in tris–HCl buffer (0.01 M, pH 7.2). The ratio of absorbance of Ct-DNA in buffer at 260 and 280 nm is of about 1.9:1 indicating that DNA was apparently free from protein contamination [31]. The concentration of DNA in the stock solution was estimated from its absorption intensity at 260 nm using a molar absorption coefficient ε260 = 6600 L mol−1 cm−1 [32]. An equal amount of DNA was added to both the reference and the test solutions in order to exclude the absorbance of DNA. The absorption titrations were carried out by varying the concentration of the DNA (5.1−6.6 × 10−5) while fixing the compound concentration at 2.8 × 10−4 M. All solutions were allowed to equilibrate thermally at 25 °C for about 10 min before the measurements.
Emission Titrations and EB Displacement Assay
Fluorescence spectroscopy has been widely employed to determine the binding propensity of small molecules with DNA. The fluoremetric experiments were conducted at a fixed concentration of the compounds (1.5 × 10−5 M) by varying the concentration of DNA (3.5−5.5 × 10−6) in tris–HCl/NaCl buffer (pH = 7.2). Before the measurements, mixtures of DNA and the test solutions were allowed to incubate for 30 min. Ethidium bromide (EB) displacement titrations were also carried out in order to find out the extent of binding of these compounds to Ct-DNA. The emission spectra of EB–DNA system in the absence and presence of each compound were recorded for the test compounds (3a-3c). The fluorescence emission intensity of EB bound to Ct-DNA was monitored in the wavelength range of 550–650 nm after exiting at 510 nm. The emission titrations were carried out by varying the sample concentration (6.1−7.5 × 10−6 M) while fixing the DNA concentration at 1.0 × 10−6 M, pretreated with ethidium bromide (1.5 × 10−6 M).
Time-Resolved Fluorescence Studies
Fluorescence lifetime measurements were made with FL920 fluorescence lifetime spectrometer (Edinburgh Instruments, Livingston, UK) operating in the time-correlated single photon counting (TCSPC) mode. All spectroscopic titration experiments, were performed at room temperature (25 °C) using a quartz cuvette of 10 mm optical path length. The samples were excited at 340 nm. The instrument response function, determined by scattering the excitation beam from a dilute suspension of colloidal silica (Ludox). The adequacy of the fits was assessed on the basis of reduced χ2 statistic and the corresponding residual distribution. The time resolved decay curves were fitted according to following tri-exponential function:
The average fluorescence lifetime was measured by using the following equation [33]:
Circular Dichroic Studies
Circular dichroic spectroscopy is one of the convenient tools for monitoring changes in DNA morphology during drug-DNA interactions. CD spectra were recorded on a Chirascan CD spectropolarimeter using a rectangular quartz cuvette of path length 1 cm. The spectra of Ct-DNA (50 mM) in the absence and presence of the test compounds were recorded in the range of 200–260 nm. All experiments were done at ambient temperature (300 K) in the 5 mM phosphate buffer solution (pH-7.2) and CD spectrum was collected after averaging three scans using a scan speed of 100 nm/min.
Viscosity Measurements
Since hydrodynamic measurementsare quite sensitive to length change, viscosity measurements are considered as most critical test of DNA binding studies. The viscosity of Ct-DNA in the absence and presence of the target compounds in the 5 mM tris–HCl/ 50 mM NaCl buffer solution (pH = 7.2) was measured using Ostwald capillary viscometer maintained at 25 ± 0.1 °C. Viscosity experiments were performed by using constant concentration of DNA while gradually increasing the concentration of test samples. Data were presented as (η/ηo)1/3 versus the ratio of the concentration of the compound to Ct-DNA, where η is the viscosity of Ct-DNA in the presence of the compound and ηo is the viscosity of Ct-DNA alone. Relative viscosity values were calculated from the observed flow time of DNA solution (t) corrected for the flow time of buffer alone (t0), using the expression ηo = (t − t0)/t0 [34].
Electrochemical Studies
The electrochemical studies of the target compounds were carried out in the absence and presence of DNA in tris-buffer (pH 7.5) and NaCl (200 mM) as supporting electrolyte. All the experiments were recorded at room temperature with three electrodes in electrochemical cell consisted of a platinum wire working electrode, a platinum counter electrode and saturated calomel electrode as reference electrode. The electrode surfaces were freshly polished with alumina powder and the solution was deoxygenated with nitrogen gas for 20 min prior to experiments. The redox behavior of the compounds (3a-3c) was checked in the absence and presence of Ct-DNA at a scan rate 0.2 Vs−1 in the potential range +1.2 to −2.0 V.
Molecular Docking Studies
Molecular docking experiments of compounds 3a-3c with B-DNAs were accomplished, in order to identify the binding affinity and interaction mode of these compounds. The crystal structures of DNA dodecamer d(CGCGAATTCGCG)2 (PDB ID: 1BNA) was extracted from protein data bank. The ligand structures for docking were prepared using ChemDraw Ultra 8.0 assigned with proper 2D orientation (ChemOffice package) and were converted to energy minimized 3D structures for in silico protein–ligand docking by “Auto-Dock Tools”. For the automated molecular docking simulations, the advanced molecular docking program AutoDock 4.0 was applied which uses a powerful Lamarckian genetic algorithm (LGA) method for conformational search and docking. The docking parameters including free binding energy, intermolecular energy and torsional free energy were calculated. Finally, the optimized geometry of test compounds with minimum binding energy was obtained.
Antimicrobial Screening
Antimicrobial screening of the compounds 3a-3c was determined against Escherichia coli (MTCC-739), Staphylococcus aureus (MTCC-737), Klebsiella pneumoniae (MTCC-109) and Salmonella typhimurium (MTCC-98) by standard micro broth dilution assay as per NCCLS protocol. MIC determination was planned using a micro dilution assay according to the Clinical and Laboratory Standards Institute Standard. The bacteria were freshly cultivated in Mueller Hinton broth at 37 °C in an incubator for 24 h and added to a 96-well plate at a final concentration of 1.0 × 105 (CFU/mL). A serial two fold dilution pattern was set in the medium wells with the concentration of compounds ranging from 1000 to 7.8125 μg/mL. A number of incongruent serial dilutions were added to each well. DMSO (10 %) with no growth inhibition of bacteria in the medium well was taken as the negative control. The least concentration of the drug was showing complete inhibition (i.e., no turbidity in the medium wells) of the visible growth after 24 h of incubation at 37 °C represented the MIC. Following 24 h, 100 μl portions were taken from the wells, serially diluted and then spotted onto Mueller Hinton agar plates. To assess the legitimacy and precision of the results, all assays were carried out in triplicate.
DPPH Free Radical Scavenging Activity
The title compounds were evaluated for their antioxidant activity using 2,2-diphenyl-1-picrylhydrazyl (DPPH) as a scavenging agent. DPPH radical assays were carried out according to reported method [35]. Briefly, 3 mL of 0.1 mM DPPH• (2,2-diphenyl-1-picrylhydrazyl) in methanol was added to 100 μl of samples (1–8 mg/mL) or ascorbic acid (0.1–0.8 mg/mL) in methanol. The above reaction mixture was incubated at 37 °C for 30 min on water bath. The DPPH scavenging activity was assessed by observing the decrease in absorbance at 517 nm using UV–Vis. spectrophotometer. All the experiments were performed in triplicate and the % inhibition was calculated as follows.
Where AControl = absorbance of DPPH• in methanol without an antioxidant and ASample = absorbance of DPPH• in the presence of an antioxidant.
Hydrogen Peroxide Free Radical Scavenging Activity
Hydrogen peroxide scavenging capability of synthesized compounds was determined by standard method [36]. The reaction mixture consisted of 3.0 mL of sample with different concentrations (0.4-1.2 mg/mL) and 1.8 mL of freshly prepared H2O2 solution in phosphate buffer saline. The reaction mixture was shaken well and incubated for 10 min and then the absorbance was recorded at 230 nm. The decrease in absorbance was measured and the percentage inhibition was calculated according to the formula:
Where AB was the absorbance of blank (without compounds) and AT was the absorbance of tested samples.
Mutagenicity Assay
Mutagenicity of the title compounds was assessed using the Ames test, as depicted by Maron and Ames [37]. The compounds were tested for their mutagenicity against S. typhimurium tester strains TA98 and TA100 both in the absence and presence of S9 mixture. The positive controls used for TA98 and TA100 were 4-nitro-o-phenylenediamine (NPD) and sodium azide (SA) without metabolic activation. In contrast 2-aminofluorene was employed as positive control for TA98 and TA100 strains respectively, with S9 mixture. The concentrations of the test compounds used were 0.02, 0.1, 0.5 and 1 mg/plate respectively and each dose was dissolved in DMSO (negative control). All assays were carried out in triplicate and revertant colonies were counted after incubation for 48 h at 37 °C.
Pharmacokinetic Properties
To assess the drug-like behavior of synthesized compounds, the pharmaceutically relevant properties such as log P, topological polar surface area (TPSA), hydrogen bond donors, hydrogen bond acceptors, molecular weight, volume and % ABS were calculated online from Mol Inspiration Chemo Informatics and are given in Table 1. Additionally, the rotational bond count which specifies the flexibility of the molecule was also determined. The percentage of absorption (% ABS) was calculated according to the equation: % ABS = 109 ± 0.345 × TPSA [38]. In general, the Lipinski “Rule of Five” is used as a filter for drug-like properties which suggests that the molecules with good membrane permeability have log P ≤ 5, number of hydrogen bond acceptors ≤ 10, molecular weight ≤ 500, and number of hydrogen bond donors ≤ 5 [39]. The predicted pharmacokinetic properties indicate that the title compounds showing no violation for Lipinski rule and all the parameters are appropriate with the acceptable range. Other molecular descriptors that determine the membrane permeability and bioavailability are reduced molecular flexibility (measured by the number of rotatable bonds), volume and polar surface area [40]. All the synthesized compounds possess significant number of rotatable bonds (3–4) and as a consequence, exhibit large conformational flexibility. The calculated percentage of absorption (% ABS) of all the test compounds is in good agreement with the acceptable range from 81 to 85 %. From the data obtained, it was concluded that all the synthesized compounds comply with these rules, suggesting their potential as a drug-like molecule.
Results and Discussion
Chemistry
The IR spectra of synthesized compounds were analyzed in the range 4000–400 cm−1. In the IR spectra of compound (2), a sharp peak appeared at 3312 and 3238 cm−1 respectively were assigned to ʋ(N-H, 1° amine) vibrations, disappeared in the compounds 3a-3c due to the condensation of amino group with respective aldehydes (see supporting information, Fig. S1). Besides, the strong absorption peak observed in the range 1626–1640 cm−1 corresponding to azomethine group indicate the formation of -CH = N bond. The stretching vibrations detected in the range 1707–1823 cm−1, were assigned to carbonyl ʋ(C = O) vibration of the imide group. However, the peaks corresponding to -OH stretching frequencies for compounds 3a, 3b were found at 3253 and 3260 cm−1.
1H NMR spectrum of compound 2 exhibited a sharp, signal at δ 5.77 ppm corresponding to NH2 proton (Fig. S2). In addition, appearance of signal corresponding to azomethine group at the range of δ 8.59–9.75 ppm confirms the formation of Schiff base. The aromatic protons appeared as multiple signals between 7.86 and 8.54 ppm respectively. The signal at δ 3.87 ppm range is assigned to –OCH3 group while the signal at δ 10 ppm range is attributed to phenolic group (−OH) of compounds 3a and 3b (Fig. 2).

1H NMR spectra of synthesized compounds (3a, 3b and 3c)
13C NMR spectra of synthesized compounds revealed signals due to carbonyl group at 160 ppm region, respectively. Signals corresponding to azomethine carbon appeared between 150.84 and 151.94 indicates the formation of proposed Schiff base framework. The multiple signals appeared between d 108 and 134 ppm is attributed to aromatic ring carbon atoms. Moreover, the signals obtained at 56 ppm range due to –OCH3 group.
The electronic spectrum of the compounds 3a-3c exhibited intense absorptions at 340 nm, which is assigned to the n-π* transition of C = N group while the bands appeared at 226 and 241 nm region attributed to π-π* energy transitions of C = O and C = N functional groups.
DNA Binding Studies
Absorbance Titrations
To explore the nature of the binding with DNA, absorbance titrations have been done for the compounds (3a-3c). The electronic absorption spectral changes of 3a-3c in the absence and presence of Ct-DNA are illustrated in Fig. 3. Upon progressive addition of Ct-DNA over a range 5.1−6.6 × 10−5 M to the fixed amount of compound concentration 2.8 × 10−4 M, the absorption bands of the title compounds exhibited hypochromism without significant changes in shape and band position. The results obtained from the UV titration experiments indicating non-covalently intercalative binding of compounds to DNA helix. In particular, hypochromism mainly arises due to intercalative mode of binding involving a strong stacking interaction between the aromatic chromophore of the compounds and the base pairs of DNA [41], whereas hyperchromism derives from the secondary damage of DNA double helix structure [42]. The observed hypochromism could be caused by the planar aromatic group of the compounds stacks between adjacent base pairs of double helix DNA.

UV–vis. absorption spectra of compounds 3a-3c (2.8 × 10−4 M) in the presence of increasing amounts of DNA (5.1−6.6 × 10−5 M). The arrow indicates the absorbance change upon increasing [DNA]. The inset is plot of DNA concentration/ (εa -εf) vs [DNA] for the titration of DNA to compounds
To compare the DNA binding strength of these compounds, the intrinsic binding constant K b was calculated by using the following host guest equation [43]:
Where, [DNA] is the concentration of DNA in base pairs, εa is the extinction coefficient observed for the compound at the given DNA concentration, εf is the extinction coefficient of the compound free in solution, and εb is the extinction coefficient of the compound when fully bound to DNA. A plot of [DNA]/ (εa–εf) versus [DNA] gave a slope 1/(εb – εf) and Y intercept equal to 1/K b (εb–εf) respectively. The intrinsic binding constant K b is the ratio of slope to intercept.
The intrinsic binding constants K b are found to be 2.25 × 104, 3.33 × 104 and 2 × 104 M−1, respectively, for the compounds (3a-3c). The decreased binding strength, which may depend on its affinity to bind DNA, follows order of 3b > 3a > 3c. The magnitude of the binding constant values indicate that the compound 3b binds more strongly to Ct-DNA than the compounds 3a and 3c.
Emission Titrations
In order to depict the binding properties of the test compounds to the Ct-DNA, fluoremetric measurements were carried out. The fluorescence emission spectra of test compounds in the absence and presence of DNA are shown in Fig. S3. The addition of increasing amounts of Ct-DNA over a range 3.5−5.5 × 10−6 M to the fixed amount of compound concentration 1.5 × 10−5 M resulted in the enhancement of the emission intensities of presented compounds, which indicated that the test compounds strongly interacted with DNA. The results obtained from the fluorimetric titrations suggest that the compounds (3a-3c) get into a hydrophobic environment inside the DNA helix and thus preventing the quenching effect of solvent water molecules [44]. The extent of hypochromism follow the order of 3b > 3a > 3c, indicating that compound 3b binds more strongly to Ct-DNA than the compounds 3a and 3c.
EB Displacement Assay
A competitive binding experiment was carried out between Ethidium bromide and the synthesized compounds with DNA to investigate the affinity of these compounds for DNA. Ethidium bromide has been extensively studied as a standard intercalating agent of DNA and it is a valuable tool to measure the magnitude of DNA binding for compounds. The fluorescence quenching of DNA bound EB by the compounds (3a-3c) are shown in Fig. 4. The fluorescence intensity of EB decreases gradually with increasing concentration of the compounds, which is an indication of competitive binding of title compounds with EB to bind to DNA. The extent of emission quenching gives a measure to determine the extent of binding among these compounds with DNA [45]. In order to quantitatively evaluate the extent of interaction, classical Sterne-Volmer equation was employed:

Emission spectra of EB bound DNA in the absence and presence of the compounds 3a-3c (6.1−7.5 × 10−5M); [EB] = 1 μM; [DNA] = 1.5 μM
Where I0 and I represent the fluorescence intensities in the absence and presence of compounds respectively, [Q] is the concentration ratio of compounds to DNA. K SV is Sterne-Volmer constant, used to evaluate the quenching efficiency and is obtained as the slope of I0/I vs [Q]. The K SV values of the compounds 3a, 3b and 3c are 1.40 × 104, 1.75 × 104 and 1.35 × 104 respectively, indicating the stronger affinity of these compounds to Ct-DNA. Thus, the data of K SV present the order of 3b > 3a > 3c, correlate well with the previous absorption spectral conclusions. Therefore, the results obtained from the competitive binding experiments provide a further evidence for the intercalation of these compounds into the DNA base stacks.
Time Resolved Fluorescence Studies
The emission decays of synthesized compounds in the absence and presence of Ct-DNA were illustrated in Fig. 5, and the calculated fitting parameters are listed in Table 2 . In the absence of Ct-DNA, compounds 3a-3c exhibits a bi-exponential decay, indicating the presence of local and polar excited states. Upon successive addition of Ct-DNA, the time resolved emission decays considerably turn from a bi-exponential pattern to a tri-exponential function. It was observed that the relative amplitude of the shorter lifetime of the probe molecules gradually increases with successive addition of Ct-DNA. In addition, there was decrease in the relative amplitude of the longer lifetime species. For example, in the case of compound 2b, we found an increase in the amplitude of shorter time component (α1) from 81 to 95 % and decrease from 19 to 4 % for longer time species (α2) upon addition of DNA. The observed enhancement in the average lifetime of test compounds might be due to the variation in the bulk viscosity of the solution with the addition of Ct-DNA [46]. Thus, the time resolved decays further confirms the interaction between the test compounds and Ct-DNA.

Time resolved fluorescence decay of compounds 3a-3c, in the absence and presence of Ct-DNA
Circular Dichoric Studies
The CD technique is a valuable method to investigate the conformational changes in the DNA, provides a useful complement to the previously used studies. In general the CD spectra of double helical right-handed B-DNA exhibits a positive band at 275 nm due to base stacking and a negative band at 240 nm due to helicity, whereas A-DNA is characterized by a positive band at 260 nm and a negative band at 210 nm in CD spectrum [47]. Simple electrostatic or groove binding interaction of compounds with DNA shows less or no perturbation of the base stacking and helicity bands, while the intensities of both the positive and negative bands affected in case of intercalative mode of binding. The CD spectra of Ct-DNA in the presence of test compounds showed significant shift in negative band position from 240 to 215 nm and a slight shift at positive band (Fig. 6). The results demonstrate that the interaction is through preferentially intercalative binding with Ct-DNA and induce conformational changes from B–DNA to A–DNA [48]. Moreover, the change induced by compound 3b was more significant than 3a and 3c which reveals that 3b has higher affinity for Ct- DNA.

CD spectra of Ct-DNA (50 mM) in the absence and presence of the compounds 3a-3c (50 mM) in 5 mM phosphate buffer
Viscosity Measurements
Further support for interaction mode between test compounds and Ct-DNA is given through the viscosity measurements. Since hydrodynamic measurements are sensitive to change in the length of DNA, viscosity study is considered as one of the most valuable techniques carried out in the absence of crystallographic structural data or NMR spectra [49]. A classical intercalation model involves the insertion of a planar molecule between DNA base pairs causes the elongation and/or rigidification of the DNA helix in solution, leading to increase in the specific viscosity [50]. On the other hand, partial and/or non-classic intercalation causes a bend or kink in the DNA helix resulting reduction in its effective length, thereby decrease in viscosity. On the addition of increasing amounts of the compound concentration to the fixed amount of DNA the viscosity of Ct-DNA is remarkably increased, suggesting that the complexes bind to DNA through an intercalation (Fig. 7). The results are in good agreement with the classical intercalators. The increase in relative viscosity associated with the compound DNA-intercalating potential, and followed the order of 3b > 3a > 3c.

Effect of increasing amounts of test compounds on the relative viscosity of DNA at pH 7.4 and 25 °C, [DNA] = 3.54 × 10−5 M and [compound] = (0.25–1.0 mM)
Electrochemical Studies
In order to further investigate the binding mode of the presented compounds with DNA, electrochemical studies have also been performed. The typical cyclic voltammograms (CV) of target compounds in the absence and presence of Ct-DNA are shown in Fig. S4. Upon the addition of DNA, the peak potential is shifted in the positive direction and the peak current is dropped by 23.3 % and 24 % for compound 3a, 3b and 22.7 % for compound 3c. The decrease in peak current is much higher for 3b than for others, suggesting its stronger DNA binding (Fig. 8). In particular, the diminution in peak current is attributed due the formation of the slowly diffusing drug–DNA supra molecular complex which leads to a decrease in concentration of the free compound in solution [51]. It has been well documented that the positive shift in peak potential is caused by the intercalation of the drug into the double helical structure of DNA, whereas negative shift is observed for the electrostatic interaction of the cationic drug-DNA backbone [52]. Therefore, the positive peak potential shift in the CV behavior of the test compounds can be attributed to the intercalation of napthalimide into the DNA double-helix correlated well with the aforementioned findings.

Cyclic voltammograms of 4.4 × 10−4 M of compound 3b in 50 mM tris-buffer, pH 7.5 at 50 mV s−1 scan rate without DNA (black) and with DNA (red)
Molecular Docking Studies
Molecular docking is an enormously useful technique, to depict the preferred orientation of drug molecule with the selected receptor. In this work, in silico studies were performed using Auto Dock 4.0 to clarify the interaction mechanism between test compounds and DNA. The docked poses of energetically most favorable conformation of compounds (3a-3c) are shown in Fig. 9 and the summary of docking results was reported in Table 3. As depicted in the Fig. 9, compound 3b interacts with the deoxy adenine18 by forming an H-bond between the CO moiety and O residue with a bond length of 2.80°A. The compound 3b also interacts with the deoxy guanine12 residue forming H-bond between the O atoms of the phosphate group of the DNA and OH moiety of 3b with bond length of 2.73°A respectively. Similarly, compounds 3a and 3c were also inserted between the planar bases of DNA and exhibited strong intermolecular hydrogen bonding interactions with the DNA bases (Fig. S5). The resulting minimum binding energies of the docked compounds–DNA complex were found to be −8.61,−8.69 and −8.20 kcal/mol for compounds 3a, 3b and 3c respectively. The more negative value of the relative binding energy indicated a greater binding potential of the compound 3b with DNA, which is consistent with the in vitro DNA binding experiments. Therefore, we conclude that there is a mutual harmonization between in vitro and in silico studies which can further validate our experimental results and afford evidence of intercalative binding.

Molecular docked model of the most favorable binding site of compounds 3b with DNA dodecamer duplex of sequence d (CGCGAATTCGCG)2 (PDB ID: 1BNA)
Antimicrobial Screening
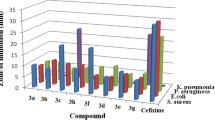
The antimicrobial profiles of the synthesized compounds were studied against Escherichia coli (MTCC-739), Staphylococcus aureus (MTCC-737), Klebsiella pneumoniae (MTCC-109) and Salmonella typhimurium (MTCC-98) by standard micro broth dilution assay as per NCCLS protocol as given in Table 4. A comparison of antibacterial activity among all the screened compounds revealed that compound 3c was found to be very promising against all the four tested bacterial strains with MIC value of 0.031 mg/mL against Escherichia coli and MIC value of 0.062 mg/mL against Salmonella typhimurium and Staphylococcus aureus. Compound 3b was found to inhibit Klebsiella pneumoniae at the MIC value of 0.062 mg/mL while both Escherichia coli and Salmonella typhimurium were moderately inhibited at the MIC value of 0.125 mg/mL. Moreover, compound 3a was found to exhibit a significant antibacterial potential against Escherichia coli, with MIC as low as 0.062 mg/mL while it showed moderate activity against Staphylococcus aureus at the MIC value of 0.125 mg/mL. The presence of additional methoxy group in compound 3c may enhance its lipophilic character, which enhances the transportation of the compound into the lipid membrane and restricts further growth of the microorganism.
DPPH Free Radical Scavenging Activity
The radical scavenging capabilities of presented compounds against stable radical DPPH were measured using UV–Vis. spectrophotometer. A significant color change from purple to yellow is the indication of reduction. The reduction capacity of DPPH radical by test compounds was determined by the decrease in its absorbance at 516 nm [53]. The results were analyzed by means of IC50 values (concentration in mg required for 50 % inhibition of DPPH radical), which are listed in Table 4. It was observed that compound 3b has highest DPPH radical scavenging activity with IC50 at 1.28 ± 0.07 mg/mL followed by 3a with IC50 at 1.43 ± 0.10 mg/mL. The investigation of antioxidant assay demonstrates that the compounds 3a and 3b showed the greater rate of DPPH scavenging activity probably due to the presence of phenolic group whereas compound 3c with electron donating methoxy group displayed moderate activity as compared to the standard ascorbic acid.
Hydrogen Peroxide Free Radical Scavenging Activity
The synthesized compounds were further evaluated for their hydrogen peroxide scavenging capabilities according to reported procedure [36]. The abilities of title compounds to scavenge the hydrogen peroxide were monitored using UV–Vis. spectrophotometer. The IC50 values of hydroxyl radical scavenging activity for the compounds 3a, 3b and 3c are found to be 1.17 ± 0.09, 1.06 ± 0.15 and 1.28 ± 0.10 mg/mL respectively (Fig. 10). In the present study, among all the compounds screened, compound 3b displayed significantly higher inhibition percentage and the results were correlated with results obtained by DPPH free radical method. The investigation of SAR studies reveals that compound 3a and 3b with OH group at para position showed greater activity whereas compounds 3c with electron donating methoxy groups exhibited moderate activity as compared to the standard ascorbic acid.

DPPH (a) and H2O2 (b) radical scavenging activities of compounds 3a-3c
Mutagenicity Assay
The title compounds were investigated for mutagenic activity using the Ames S. typhimurium reversion assay. Obtained data is shown in Table 5 which represents the mean number of revertants/plate after the treatment of test compounds with S. typhimurium strains TA98 and TA100, respectively with and without metabolic activation. It was observed that the test compounds had no mutagenic activity in TA98 and TA98 strains both in the absence and presence of S9 mix. In general, a significant increase in the number of revertant colonies compared to the solvent control is considered as the indication of mutagenic activity of test samples [54]. Since there were no two-fold increase in colony numbers of negative control observed, all the test compound were found to be non-mutagenic in the presence and absence of metabolic activation.
Conclusions
In this article, we report the synthesis and biophysical evaluation of a new series of N-substituted naphthalimide derivatives as promising DNA binding, antioxidant and antimicrobial agents. DNA binding studies were explored by employing hydrodynamic, electrochemical and multi-spectroscopic techniques and the findings suggested that the test compounds interact with DNA via intercalative binding. The displacement of EB by compounds (3a-3c), viscosity changes, positive shift in peak potential and conformational changes from B–DNA to A–DNA were collectively provide evidences for the intercalation of these compounds into the DNA base stacks. The magnitude of the binding and Sterne-Volmer constant values of these compounds follows the order of 3b > 3a > 3c, indicating that the compound 3b binds more strongly to Ct-DNA than the compounds (3a and 3c). Moreover, the lower binding free energy of 3b compared to 3a and 3c demonstrated that the DNA-binding propensity of the compound 3b is stronger than the compound 3a and 3c. All the compounds were subjected for antimicrobial screening and compound 3c with additional methoxy group, emerged as potential agent against all the selected bacterial strains. The results obtained from mutagenic assay demonstrates that the test compounds were non-mutagenic in nature. Furthermore, the investigation of antioxidant assay reveals that the compounds 3a and 3b showed the greater rate of DPPH and H2O2 scavenging activities possibly due to the presence of phenolic group at para position.
DNA, Deoxyribonucleic acid; Ct-DNA, Calf-thymus DNA; CD, Circular dichroism; DMSO, Dimethyl sulfoxide; SCE, Saturated calomel electrode; MIC, Minimum inhibitory concentration.
References
Chen Z, Liang X, Zhang H, Xie H, Liu J, Xu Y, Zhu W, Wang Y, Wang X, Tan S, Kuang D, Qian X (2010) A new class of naphthalimide-based antitumor agents that inhibit Topoisomerase II and induce lysosomal membrane permeabilization and apoptosis. J Med Chem 53:2589–2600
Fuente RDL, Sonawane ND, Arumainayagam D, Verkman AS (2006) Small molecules with antimicrobial activity against E. coli and P. aeruginosa identified by throughput screening. Br J Pharmacol 49:551–559
Tischer M, Sologub L, Pradel G, Holzgrabe U (2010) The bisnaphthalimides as new active lead compounds against Plasmodium falciparum. Bioorg Med Chem 18:2998–3003
Xu Y, Qian X, Yao W, Mao P, Cui J (2003) Novel naphthalimide hydroperoxide Photonucleases: the role of Thiocyclic-fused area and the difference in spectra, photochemistry and photobiological activity. Bioorg Med Chem 11:5427–5433
Qian X, Li Y, Xu Y, Liu Y, Qu B (2004) Highly-efficient DNA photocleavers with long wavelength absorptions: thio-heterocyclic fused naphthalimides containing aminoalkyl side chains. Bioorg Med Chem Lett 14:2665–2668
Ingrassia L, Lefranc F, Kiss R, Mijatovic T (2009) Naphthalimides and azonafides as promising anti-cancer agents. Curr Med Chem 16:1192–1213
Gregor KAM, Robertson MJ, Young KA, Kleist LV, Stahlschmidt W, Whiting A, Chau N, Robinson PJ, Haucke V, Mc Cluskey A (2014) Development of 1, 8-Naphthalimides as Clathrin Inhibitors. J Med Chem 57:131–143
Cao H, Chang V, Hernandez R, Heagy MD (2005) Matrix screening of substituted N-Aryl-1,8-naphthalimides reveals new dual fluorescent dyes and unusually bright pyridine derivatives. J Org Chem 70:4929–4934
Duke RM, Gunlaugsson T (2007) Selective fluorescent PET sensing of fluoride (F) using naphthalimide–thiourea and –urea conjugates. Tetrahedron Lett 48:8043–8047
Gan J, Song QL, Hou XY, Chen K, Tian H (2004) 1,8-Naphthalimides for non-doping OLEDs: the tunable emission color from blue, green to red. J Photochem Photobiol A Chem 162:399–406
Zhang YY, Zhou CH (2011) Synthesis and activities of naphthalimide azoles as a new type of antibacterial and antifungal agents. Bioorg Med Chem Lett 21:4349–4352
Brana MF, Cacho M, Garcia MA, Pascual-Teresa B, Ramos A, Dominguez MT, Pozuelo JM, Abradelo C, Rey-Stolle MF, Yuste M, Banez-Coronel M, Lacal JC (2004) New analogues of amonafide and elinafide, containing aromatic heterocycles: Synthesis, antitumor activity, molecular modeling, and DNA binding properties. J Med Chem 47:1391–1399
Li Y, Yang ZY, Wang MF (2009) Synthesis, characterization, DNA binding properties and antioxidant activity of Ln(III) complexes with hesperetin-4-one-(benzoyl) hydrazone. Eur J Med Chem 44:4585–4595
Kashanian S, Javanmardi S, Chitsazan A, Paknejad M, Omidfar K (2012) Fluorometric study of fluoxetine DNA binding. J Photochem Photobiol B 113:1–6
Tong C, Xiang G, Bai Y (2010) Interaction of paraquat with Calf thymus DNA: a Terbium (III) luminescent probe and multispectral study. J Agric Food Chem 58:5257–5262
Guo H, Cai C, Gong H, Chen X (2011) Multi-spectroscopic method study the interaction of anti-inflammatory drug ketoprofen and calf thymus DNA and its analytical application. Spectrochimic Acta Part A A79:92–96
Kalanur SS, Katrahalli U, Seetharamappa J (2009) Electrochemical studies and spectroscopic investigations on the interaction of an anticancer drug with DNA and their analytical applications. J Electroanal Chem 636:93–100
Oehlers L, Mazzitelli CL, Brodbelt JS, Rodriguez M, Kerwin S (2004) Evaluation of complexes of DNA duplexes and novel benzoxazoles or benzimidazoles by Electro spray ionization mass spectrometry. J Am Soc Mass Spectrom 15:1593–1603
Jangir DK, Charak S, Mehrotra R, Kundu S (2011) FTIR and circular dichroism spectroscopic study of interaction of 5-fluorouracil with DNA. J Photochem Photobiol B 105:143–148
Cusumano M, Pietro MLD, Giannetto A, Nicolo F, Rotondo E (1998) Noncovalent interactions of Platinum (II) square planar complexes containing ligands out-of-plane with DNA. Inorg Chem 37:563–568
Vijesh AM, Isloor AM, Telkar S, Arulmoli T, Fun HK (2013) Molecular docking studies of new imidazole derivatives for antimicrobial properties. Arabian J Chem 6:197–204
Xie L, Xu Y, Wang F, Liu J, Qian X, Cui J (2009) Synthesis of new amonafide analogues via coupling reaction and their cytotoxic evaluation and DNA-binding studies. Bioorg Med Chem 17:804–810
Zhao L, Li J, Li Y, Liu J, Wirth T, Li Z (2012) Selenium-containing naphthalimides as anticancer agents: design, synthesis and bioactivity. Bioorg Med Chem 20:2558–2563
Mortelmans K, Zeiger E (2000) The Ames Salmonella/microsome mutagenicity assay. Mutat Res 455:29–60
Arriaga-Alba M, Ruiz-Pérez NJ, Sánchez-Navarrete J, López de Angel B, Flores-Lozada J, Blasco JL (2013) Antimutagenic evaluation of vitamins B1, B6 and B12 in vitro and in vivo, with the Ames test. Food Chem Toxicolol 53:228–234
Vicini P, Geronikaki A, Incerti M, Busonera B, Poni G, Cabras CA, Colla PL (2003) Synthesis and biological evaluation of Benzo[d]isothiazole, Benzothiazole and Thiazole Schiff bases. Bioorg Med Chem 11:4785–4789
Cheng K, Zheng QZ, Hou J, Zhou Y, Liu CH, Zhao J, Zhu HL (2010) Synthesis, molecular modeling and biological evaluation of PSB as targeted antibiotics. Bioorg Med Chem 18:2447–2455
Karthikeyan MS, Prasad DJ, Poojary B, Bhat KS, Holla BS, Kumari NS (2006) Synthesis and biological activity of Schiff and mannich bases bearing 2, 4-dichloro-5-fluorophenyl moiety. Bioorg Med Chem 14:7482–7489
Kumar KS, Ganguly S, Veerasamy R, Clercq ED (2010) Synthesis, antiviral activity and cytotoxicity evaluation of Schiff bases of some 2-phenyl quinazoline-4(3)H-ones. Eur J Med Chem 5:5474–5479
Pu-Su Z, Jie S, Xiao-Jun S, Li D, Fang-Fang J (2011) Syntheses, crystal structures and property studies on Cu(II) and Co(II) complexes with 1,8-Naphthalimide Schiff base Chinese. J Struct Chem 30:346–353
Arish D, Nair MS (2011) Synthesis, spectroscopic, antimicrobial, DNA binding and cleavage studies of some metal complexes involving symmetrical bidentate N, N donor Schiff base ligand. Spectrochim Acta A 82:191–199
Anbu S, Kandaswamy M (2011) Electrochemical, magnetic, catalytic, DNA binding and cleavage studies of new mono and binuclear copper(II) complexes. Polyhedron 30:123–131
Ghosh S, Banik D, Roy A, Kundu N, Kuchlyan J, Sarkar N (2014) Spectroscopic investigation of the binding interactions of a membrane potential molecule in various supra molecular confined environments: contrasting behavior of surfactant molecules in relocation or release of the probe between nano carriers and DNA surface. Phys Chem Chem Phys 16:25024–25038
Rajendiran V, Murali M, Suresh E, Palaniandavar M, Periasamy VS, Akbarsha MA (2008) Non-covalent DNA binding and cytotoxicity of certain mixed-ligand ruthenium(II) complexes of 2,2 -dipyridylamine and diimines. Dalton Trans. 2157–2170
Miliauskas G, Venskutonis PR, Beek TAV (2004) Screening of radical scavenging activity of some medicinal and aromatic plant extract. Food Chem 85:231–237
Ruch RJ, Cheng SJ, Klaininig JE (1980) Prevention of cytotoxicity and inhibition of intracellular communication by antioxidant catechins isolated from Chinese green tea. Carcinogen 10:1003–1008
Maron DM, Ames BN (1983) Revised methods for Salmonella mutagenicity test. Mutat Res 113:175–215
Wang R, Fu Y, Lai L (1997) A new atom-additive method for calculating partition coefficients. J Chem Inf Comput Sci 37:615–621
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development setting. Adv Drug Deliv Rev 23:3–25
Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kapple KD (2002) Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem 45:2615–2323
Wolfe A, Shimer GH, Meehan T (1987) Polycyclic Aromatic hydrocarbons physically intercalate into duplex regions of denatured DNA. Biochemistry 26:6392–6396
Barton JK, Danishefsky AT, Goldberg JM (1984) Tris(phenanthroline) ruthenium(II): stereo selectivity in binding to DNA. J Am Chem Soc 106:2172–2176
Shah A, Zaheer M, Qureshi R, Akhter Z, Nazar MF (2010) Voltammetric and spectroscopic investigations of 4-nitrophenylferrocene interacting with DNA. Spectrochim Acta A 75:1082–1087
Arjmand F, Muddassir M, Yousuf I (2014) Design and synthesis of enantiomeric (R) and (S)-copper (II) and diorganotin(IV)-based antitumor agents: Their in vitro DNA binding profile, cleavage efficiency and cytotoxicity studies. J Photochem Photobiol B 136:62–71
Arjmand F, Yousuf I, Hadda T, Toupet L (2014) Synthesis, crystal structure and anti proliferative activity of Cu(II) nalidixicacide DACH conjugate: Comparative in vitro DNA/RNA binding profile, cleavage activity and molecular docking studies. Eur J Med Chem 81:76–88
Sowrirajan C, Yousuf S, Vijayaraj MVEI (2014) Binding of flavanone with-CD/ct-DNA: a spectroscopic investigation. Turk J Chem 38:725–738
Kypr J, Kejnovska I, Renciuk D, Vorlıckova M (2009) Circular dichroism and conformational polymorphism of DNA. Nucleic Acids Res 37:1713–1725
Jose D, Porschke D (2004) Dynamics of the B-A transition of DNA double helices. Nucleic Acids Res 32:2251–2258
Sigman DS, Mazumder A, Perrin DM (1993) Chemical nucleases. Chem Rev 93:2295–2316
Wang B, Yang ZY, Crewdson P, Wang D (2007) Synthesis, crystal structure and DNA-binding studies of the Ln (III) complex with 6-hydroxychromone-3-carbaldehyde benzoyl hydrazone. J Inorg Biochem 101:1492–1504
Chu X, Shen GL, Jiang JH, Kang TF, Xiong B, Yu RQ (1998) Voltammetric studies of the interaction of daunomycin anticancer drug with DNA and analytical applications. Anal Chim Acta 373:29–38
Aslanoglu M (2006) Electrochemical and spectroscopic studies of the interaction of Proflavine with DNA. Anal Sci 22:439–443
Latha P, Kodisundaram P, Sundararajan ML, Jeyakumar T (2014) Synthesis, characterization, crystal structure and theoretical study of a compound with benzodiazole ring: antimicrobial activity and DNA binding. Spectrochim Acta Part A 129:429–437
Erena Y, Özata A (2014) Determination of mutagenic and cytotoxic effects of Limonium globuliferum aqueous extracts by Allium, Ames, and MTT tests. Rev Bras Farmacogn 24:51–59
Acknowledgments
The authors thank to University Grant Commission, New Delhi for financial support through Major Research Project (F.No. 41-238/2012) and Mr. Pattan Sirajuddin. Nayab is also thankful to UGC for meritorious BSR Fellowship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors confirm that this article content has no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 1405 kb)
Rights and permissions
About this article

Cite this article
Nayab, P.S., Pulaganti, M., Chitta, S.K. et al. Evaluation of DNA Binding, Radicals Scavenging and Antimicrobial Studies of Newly Synthesized N-Substituted Naphthalimides: Spectroscopic and Molecular Docking Investigations. J Fluoresc 25, 1905–1920 (2015). https://doi.org/10.1007/s10895-015-1683-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10895-015-1683-1