
Abstract
XRD and TEM characterisation evidenced the formation of well-dispersed CdS nanoparticles inside a phosphate glass matrix. Optical absorption and time-resolved photoluminescence study were carried out on the prepared glass samples. Optical absorption revealed the fast character of the growth of CdS nanoparticles in this medium. Photoluminescence spectra showed only one large band with a maximum at almost 740 nm, which was associated to transitions between energy levels within the bandgap of the CdS nanoparticles. From the steady state and time-resolved measurements, it was suggested that the emission comes mainly from sulfur vacancies inside the nanocrystals and on its surface, which act as deep traps for the photogenerated electrons. The creation of such vacancies was attributed to the loss of sulfur during the glass preparation as evidenced from a chemical analysis using energy dispersive X-ray spectrometry. These traps may be also induced by the fast growth of CdS nanocrystals in this matrix or laser exposure during PL measurements. These CdS-doped glasses with an intense absorption in the UV–Vis region and a large emission band with long lifetime and a large Stokes-shift are adequate for luminescent solar concentrators, photocatalytic applications and solid-state lasers.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Semiconductor nanoparticles with size of few nanometres have physical properties largely different from that of the corresponding bulk crystal. Since the appearance of the first reports on these materials there is 30 years ago [1,2,3,4,5], the interest paid to them is still growing. A great number of theoretical and experimental studies dealing with the physical properties of these nanocrystals were carried out, and new techniques of their preparation have been developed [6,7,8,9,10,11,12,13].
The main reason, which motivates the scientific research in this field, is the investigation of the fundamental properties of these materials in addition to the possibility of their use in different technological applications [14,15,16,17].
Cadmium sulphide (CdS), a II–VI semiconductor with a direct band gap of nearly 2.4 eV, has received considerable attention because of its promising applications in optoelectronic devices, biosensors and photocatalysis. However, the use of CdS nanoparticles is often restricted due to the great density of surface defects, which appear due to dangling bonds, impurities and vacancies. Since these defects provide traps for charge carriers, they open new pathways for non-radiative recombination and strongly affect the emission. Indeed, the emission of these nanoparticles is usually composed of a narrow band-edge peak and a large band associated to trap states. In order to adjust the emission of these materials for a specific application, it is very important to control the ratio between these two bands. The enhancement of the PL emission band-edge will be obtained by the elimination of surface defects through a convenient passivation, which may be achieved by different methods [18,19,20,21,22,23,24,25,26,27,28]. Unfortunately, neither of these methods has been found to eliminate efficiently surface defects. Therefore, one must adjust its own need according to the emission properties of these nanocrystals. In particular, the red and NIR emission in the range between 700 and 1000 nm may be exploited in biology since their penetration into bio-samples can reach 2–5 cm with a weak attenuation [29]. Additionally, CdS nanoparticles providing largely shifted emission from absorption onset may provide a solution to prevent reabsorption in luminescent solar concentrators (LSC) and replace core-shell nanostructures, which are so much difficult to prepare [19]. Therefore, Stokes-shift-engineered nanocrystals undergo renewal interest for Large-area luminescent solar concentrators [30]. On the other hand, CdS is an attractive material which has been extensively used as a photocatalyst for solar hydrogen production and degradation of organic dyes [16, 22].
Semiconductor nanoparticles were grown for the first time in silicate glass hosts and they were used as coloured glasses since the antiquity. However, the high temperature processing as well as the long annealing time necessary to the growth of the nanocrystals and the difficulty of dissolving high concentrations of the semiconductor phase ins silicate glasses were considered as limiting factors for the use of such materials in different optoelectronic applications [31]. All these drawbacks have prompted the searchers to find low-melting glasses, which can dissolve higher concentrations of semiconductor. In this context, phosphate glasses are known to have low glass transition and processing temperatures. Additionally, they can host relatively high amounts of transition metal ions as well as rare earth elements without loss of their main properties. Therefore, these materials appeared to offer the possibility of low temperature annealing and the dispersion of higher quantity of semiconductor as compared to silicate glasses. However, it took long time before seeing the first report on the growth of CdS nanoparticles inside a phosphate glass matrix, which appeared in the middle of 1990 [32]. It was demonstrated in this report, that nanocrystals growth in phosphate glasses occurs even when they are annealed at lower temperatures and for shorter durations as compared to silicate ones. The reasons behind this finding are the low glass transition temperature and viscosity of these materials [33]. So far, these results have encouraged many other groups to prepare different phosphate glass hosts putting as main objective the growth of semiconductor nanocrystals with narrow size dispersion and high concentration [26, 34,35,36,37,38,39]. Indeed, the temperature of dissolution and the composition of the glass matrix are considered crucial parameters to dissolve higher semiconductor concentrations and to control the growth kinetic and the size distribution of the nanocrystals [33]. On the other hand, pure phosphate glasses suffer from a low chemical durability due to the presence of hygroscopic P–O–P bonds, which limits their use in many applications. One way to improve the devitrification resistance and the chemical durability is the addition of heavy metal oxides. In particular, the addition of barium oxide (BaO) to phosphate has several benefic effects by increasing the chemical durability, the devitrification resistance and the refractive index of the glass [40]. Therefore, phosphate glasses equipped with such excellent properties may be good candidates for many applications. In particular, glasses suitably doped with semiconductor nanoparticles, which present large Stokes-shift, are highly researched to enhance the efficiency of LSC by diminishing the noxious effects of light reabsorption [15, 19]. Moreover, to overcome photocorrosion effect, CdS nanoparticles embedded in a glass matrix were proposed as efficient recyclable photocatalyst [14].
Accordingly, we present in this paper the results of our study on a barium phosphate glass doped with cadmium sulphide (CdS) nanoparticles by the use XRD, TEM, optical absorption and photoluminescence (PL) spectroscopic techniques. We will be interested by elucidating the origin of the large Stokes-shift and the nature of the states involved in emission of such nanoparticles. This is necessary for the design, optimization, and development of optoelectronic devices based on these glasses.
Experimentals
Glass Preparation
The glass subject of this study was prepared in the following composition, 57 mol% of P2O5, 28.5 mol% of BaO and 14.5 mol% of K2O, by melting the starting reagents in a platinum crucible, in an electric furnace heated up to 1350 °C in air atmosphere. The formed glass was then grinded to obtain a fine powder. After that, convenient weights of CdO and Na2S were added as dopants to the powder in order to obtain 1.2% W of CdS in the final composition before melting again. To minimise sulfur loss, the mixture was sealed in a fused silica tube, and heated up to 1100 °C for 15 min. The glass had been homogenized in the melt by shaking the tube for several times, and quenched by immersing carefully the tube in water to avoid fast precipitation of the semiconductor phase. The final glass prepared through this process was thus transparent and colourless. Small samples of 5 × 5 × 2 mm3 were cut and polished to obtain slices for optical measurements. Before thermal treatment to grow semiconductor nanoparticles, the glass transition temperature (T g ) and the crystallization onset temperature (T x ) were identified by performing differential scanning calorimetry analysis (DSC) and they were found to be nearly equal to 350 and 417 °C respectively. Hereafter, the samples were differently heat-treated at a temperature between T g and T x in order to get low viscosity and to initiate nucleation. The glass was gradually coloured from light yellow to dark orange depending on the time and the temperature of the treatment. This coloration occurred only for annealing temperature equal or greater than the glass transition temperature T g .
Experimental Techniques
DSC thermogram was performed on powder glass samples in sealed platinum pans under N2 atmosphere at a constant rate of 10 °C/min by using a Labsys TG-DSC16 apparatus from SETARAM. X-ray diffraction (XRD) patterns were recorded by means of an X-ray diffractometer (PANALYTICAL) using CuKα radiation of wavelength 0.15406 nm in the scanning 2θ range from 30 to 60°. For performing transmission electron microscopy (TEM), JEOL 2010 FEG apparatus was employed. Elemental chemical analysis was done using an energy dispersive X-ray spectrometer (EDAX, OXFORD instrument) coupled to a scanning electron microscope (SEM JEOL, JSM 6510 LV). Optical absorption spectra were measured at room temperature using an UV-vis Shimadzu 1200 spectrophotometer. PL emission measurements were performed using either the 355 or 532 nm wavelengths of an Nd3+: YAG pulsed laser as excitation source. The acquisition of such emitted light was achieved by using a spectrometer (Oriel, grating 400 or 1200 l/mm) coupled to a time-gated intensified CCD detector. Such equipment allows time-resolved measurements with a minimum time resolution of 4 ns (delay and gate widths).
Results and Discussions
XRD, TEM and EDX Analysis
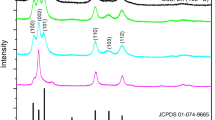
XRD analysis was used to investigate the phase structure and crystalline size of CdS nanoparticles grown inside the glass matrix after heat-treatment. Figure 1 shows the XRD pattern of the glass sample annealed at 365 °C for 10 min. This pattern exhibits clear and relatively intense diffraction peaks, which may be due to the great concentration of CdS nanocrystals or the existence of large particles with good crystallinity. By using Joint Committee on Powder Diffraction Standards (JCPDS) cards, the observed XRD peaks were assigned to either the hexagonal wurtzite or cubic zincblende crystalline phase (Table 1). These results clearly indicate that cubic and hexagonal phases coexist in the prepared glass samples. It may be pointed out that the 2θ positions of (0 0 2), (1 1 0) and (112) lines of hexagonal phase have the same 2θ values as those for (1 1 1), (2 2 0) and (311) lines of cubic phase respectively. On the other hand, the crystallite size was calculated using the Debye–Scherrer formula:
where λ is the wavelength of X-ray diffraction [λ = 0.15418 (Cu K-alpha)], β is the Full Width at Half Maximum (FWHM, in radians) of the most intense peak of the XRD pattern and θ is the angle of diffraction. From the high intense reflection peak observed at 2θ = 26.0°, the particle size in the investigated glass sample was found to be 27.3 nm.

XRD pattern for the glass sample annealed at 365 °C for 10 min
Figure 2 shows the TEM image of the glass sample heat-treated at 365 °C for 10 min. This figure clearly shows small nearly spherical CdS nanocrystals well dispersed within the glass matrix together with large nanoparticles, which resulted from aggregation and coalescence of smaller ones. The large size dispersion between small and big nanoparticles may be related to the fast growth of the semiconductor phase inside this glass matrix. By comparing XRD measurement to TEM image, we may suggest that the size obtained from XRD is related to the biggest nanoparticles since they possess more diffracting planes than smaller ones.

TEM image for the glass sample annealed at 365 °C for 10 min
Elemental chemical analysis of the glass matrix was obtained throw energy dispersive X-ray spectroscopy (EDX). The analysis of the spectra obtained at different region of the glass powder (Figs. 3, 4) are summarized in Table 2 and they clearly show a large amount of cadmium as compared with sulfur although we have initially used the same quantity for the two elements in the initial composition. In addition to the used components in the preparation of the glass, we detect the presence of aluminum, which may be introduced in the glass as impurity whereas silica may come from the quartz tube used during the melt of the glass.

SEM image of the powder obtained by grinding the glass sample annealed at 365 °C for 10 min

EDX spectrum for the glass sample heat-treated at 365 °C for 10 min
Optical Absorption Study
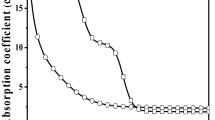
Figure 5 shows the optical spectra for differently thermal-treated glass samples. These spectra resemble those found for CdS nanocrystals in others phosphate glasses [26]. By comparing with that of the as-made hosting glass, we can observe a significant change in the spectra profiles which occurs as the time or the temperature of the heat-treatment increased. This change is usually attributed to the growth of semiconductor particles in the glass matrix. Besides, a clearly shift of the optical absorption edge toward lower energies when increasing the time or temperature of the treatment was also observed. This redshift indicates the growth of CdS nanoparticles with larger sizes, which results in the reduction of the so-called quantum size effect due to carrier confinement in the CdS nanocrystals [1, 4]. The fact that the optical spectra are without well-structured excitonic peaks may be due to several factors, among which the thermal broadening, which may be important at room temperature. Besides, big crystallites or a wide size distribution as was confirmed by XRD and transmission electron microscopy measurements may be another reason for this behavior. Additionally, internal defects and surface states introduce states that extend into the forbidden gap and contribute to the entire absorption spectra giving a redshift of the absorption onset [27].

Optical absorption spectra for the non-annealed sample (NAG) and the differently heat-treated glass samples. It is shown as inset an example of the second derivative of an optical spectrum
In order to characterize the growth process of CdS nanocrystals in this phosphate glass system, we have estimated the bandgap relative to each sample from the wavelength, which corresponds to the minimum position of the second derivative of its optical absorption spectrum [28, 41, 42]. The results of our calculations are reported in Table 3. Based on the effective-mass approximation (EMA), the optical bandgap for a spherical semiconductor nanoparticle within the dielectric glass medium can be deduced from the following equation [2, 3]:
where Eg(∞) (~ 2.40 eV) is the bandgap of bulk CdS, ħ, the Planck’s constant, μ, the reduced mass of CdS exciton (~ 0.154m e ), e and m e are the charge and the mass of the electron and ε is the CdS dielectric constant (~ 8.9).
Solving Eq. (2) for the diameter d, one obtains the following relation between the particle diameter, d, (in nm) and the experimentally observed bandgap energy E g , in electron volts:
The estimated nanocrystals diameters are reported in Table 3. The discrepancy between the particles size obtained from XRD and that estimated from the EMA model for the sample annealed at 365 °C for 10 min suggests a weak quantum confinement effect because of irregular growth of CdS nanocrystals and the presence of nanoparticles with large size. Additionally, EMA uses parameters related to the bulk material as numerical inputs, which may be another possible reason for the observed discrepancy. However, the analysis of the particle size dependence on the time and temperature of the heat-treatment, allowed us to make the following proposals to explain the evolution of the optical spectra:
-
1.
The particles grow rapidly and their mean diameter takes greater values than the Bohr exciton diameter (5.2 nm). This is supported by the absence of a clear excitonic absorption peak even for short time or low temperature of the heat-treatment. Similar tendency was observed for the growth of CdSe nanoparticles in a fluorophosphate glass matrix [43, 44].The featureless spectra approach that which are expected for large spheres. Indeed, the smaller are the crystals size, the higher the spectral resolution will be observed. Thus, we believe, that the nucleation stage in this glass system is very fast especially for high heat-treatment temperature (365 °C).
-
2.
The growth of particles during the diffusion-controlled stage occurs at the first minute of the treatment and the average particle size rapidly reaches nearly 7 nm with the duration of treatment. This is due to the low viscosity induced by the presence of potassium and sodium in the glass composition [45].
-
3.
For a temperature of 350 °C and a long annealing time (> 50 min), we observed an absorption spectrum similar to that obtained for bulk CdS. Further heat-treatment leads to an increase of the absorption without significant absorption edge spectral shift. We think thus, that after the 50th min of heat-treatment and at a temperature of 350 °C, the coalescence stage takes place resulting in particles with very large sizes. Indeed, some nanocrystals may be created close enough to each other, and even at the first stage of nanocluster growth at higher heat-treatment temperature, coalescence of some nanoclusters may occur. This suggestion seems to be in agreement with the results deduced from XRD and TEM characterisations.
It is worth to note however, that these glasses doped with CdS nanoparticles, which are easy to prepare and have suitable band gap and efficient absorption of the visible light, may be more useful to achieve solar-driven photocatalysts for hydrogen production [14].
Photoluminescence Study
PL Spectra
The PL spectra of the differently heat-treated samples obtained under an excitation wavelength of 532 nm are presented in Fig. 6. Apart from minor variations, they are nearly identical in shape and each of them shows only one large band extending into the region between 600 and 850 nm even when we used an excitation energy (λ exc = 355 nm) larger than the band gap energy of the smallest nanoparticles (Fig. 7). A shoulder around 775 nm is also observable in all the PL spectra indicating that at least two bands compose this emission. The authors of Ref. [16] observed similar results. This emission, which occurs in the red to near-IR (> 600 nm) wavelength window, is useful for laser quantum dots applications in biology. Additionally, the PL band observed in this study, with a large Stokes-shift from absorption onset, which exceeds 200 nm, is favorable for application in LSC due to the reduction of the reabsorption effects [46].

Photoluminescence spectra of the differently heat-treated glass samples under an excitation wavelength of 532 nm

Photoluminescence spectra obtained under two excitation wavelengths (355 and 532 nm) for the glass sample heat-treated at 365 °C for 5 min
In semiconductor nanocrystals, the emission can occur through intrinsic defects inside the nanoparticles due to the deviation from the ideal stoichiometry and to surface-related trap states due to unquenched dangling orbitals. Indeed, It was reported that this traps may be of different origins such as substituted impurities, lattice distortions or surface defects [47, 48]. The large bandwidth at half maximum of the emission band (~ 115 nm) evidences either a wide particle size distribution, trapping states with different energies or a strong phonon coupling of the electron traps [49]. The width remains however unchanged with nanoparticles size, which argues that emission involves deep traps [50]. In fact, such traps are more localized and their wave functions are less extended than shallow ones and free carriers. Therefore, their emission is less influenced by the change in crystallite size [49, 51]. For shallow traps, due to the strong mixing of their wave functions with valence and conduction band, the energy of the charge carrier follows the variation of the bandgap, which influences the wavelength of the emission band. Moreover, shallower traps are suggested to emit at shorter wavelengths, whereas deeper ones are expected to emit at lower energies [52]. Besides, transitions involving shallow states are usually thermally quenched because of the small activation barriers between shallow states and either the conduction band edge or valence band edge.
Even though the PL spectra of II–VI semiconductors nanocrystals were not clearly explained in literature and there is no common agreement so far established on the luminescence process, many possible schemes were proposed to explain the radiative electron-hole recombination in CdS nanoparticles. Among the different pathways, we find the transition from a sulfur vacancy (VS) or cadmium interstitial (ICd) to the valence band, from the conduction band or a sulfur interstitial (IS) to a cadmium vacancy (VCd) associated with a complex [53,54,55]. Emission may be due also to the radiative recombination between a hole bound to an ionized acceptor and an electron bound to an ionized donor, which is referred to as the “Donor-Acceptor Pair” (DAP) transition [56]. The particularity of DAP emission is its great dependence on the nanocrystals stoichiometry since crystals defect will generate more and more donor and acceptor sites. Indeed, it was suggested in the earlier literature, that sulfur vacancy and interstitial cadmium act as donor states, whereas the acceptor states arise from cadmium vacancy and interstitial sulfur. The donor states were reported to be below the conduction band in the range from 0.6 to 1.0 eV for VS, whereas they were suggested to be in the range from 0.12 to 0.26 eV for ICd. The acceptor states were believed to be above the valence band in the range from 0.26 to 1.2 eV for VCd, and between 0.11 to 0.15 eV for IS defects [57, 58]. In addition, anti-sites such as Scd and CdS may also play the role of carrier traps. Additional trap levels may be present due to the insertion of foreign atoms in the nanocrystals structure. In particular, some atomic impurities like as Na or K, which are present in the glass matrix, may diffuse inside the CdS nanocrystals during the growth.
It is reported that emissions involving only one shallow trap level (IS or ICd) falls into the spectral region below 600 nm [58]. On the other hand, transitions related to only one deep trap (VS or VCd) result in emission in the red region. Moreover, some reports have also shown that the broad defect emissions above 600 nm involve recombination of donor–acceptor pairs. These emissions may be due to transitions between cadmium interstitials and cadmium vacancy states or between sulfur vacancy and cadmium vacancy states [59, 60]. Additionally, the emission band with a maximum around 760 nm was attributed to self-activated center consisting of Cd–S composite vacancies (VCd–S), which is a donor to acceptor level transition [61]. Therefore, the energetic position of the large emission band extending from the red to the near-infrared region, observed in our spectra may contain many independent emission bands causing considerable confusion in their assignment. However, the small shift of this band with the particle size suggests that at least one type of charge carriers are localized on nearly deep traps, whereas the others are localized on either a shallow or a deep trap. Therefore, we believe that the emission band observed in this study is possibly due to the recombination of electrons and holes trapped in different types of DAPs among which, we may find transitions between VS and IS, ICd and VCd. Due to the small energetic gap, transitions between VS and VCd give rise to the emission in the red region. This suggestion seems to be in reasonable agreement with the results of Xu et al. [59], who observed a PL band at 760 nm in CdS nanoparticles, which they attributed to DAP recombination process involving holes trapped by the VCd centers and electrons trapped by the VS. According to these authors, self-activated VCd or ICd are created when S vacancy centers (VS) are present above a threshold concentration. On the other hand, Suyver [62], observed, when increasing the S2- concentration beyond the stoichiometric ratio in ZnS: Mn colloids, a decrease of the emission related to sulfur vacancies. Moreover, by contrast to our observations, it was found that CdS-doped glasses, with cadmium vacancies as the main defects, emit in the region between 550 and 650 nm [63]. Furthermore, the decrease of the emission intensity with decreasing the particle size observed in this study, suggests that this emission belongs mainly to deep-trapping states in the volume of CdS nanoparticles rather than surfaces defects [55]. Otherwise, emission intensity must increases since the surface to volume rate increases. Accordingly, we believe that CdS nanoparticles grown in our phosphate glass system suffer from a great amount of surface trapping states and internal defects. This may be due to an important loss of sulfur stemming from its high volatility and the low viscosity of phosphate glasses, which results in the formation of sulfur vacancies. This suggestion is in good agreement with the elemental chemical analysis, which show a smaller amount of sulfur in the final glass as compared to that of cadmium. Additionally, the low viscosity allows a more effective diffusion and consequently a fast particle growth leading thus to an incomplete shielding of the outer layers and the creation of a great number of defects inside the nanocrystal and on its surface due to ionic displacements during recrystallization [64, 65]. Because of the loss of sulfur, additional ICd and VCd defect levels are formed during annealing [47]. Moreover, defect states may also occur during laser exposure through photosensitivity effects [66,67,68].
Although the band position and the shape of the emission remain almost unchanged with the excitation wavelength (Fig. 7), we cannot conclude about the size distribution of crystallites. According to Chestnoy et al. [53], the emission is controlled by the nature and the density of the emitting states together with the distribution of CdS particle sizes. However, the large Stokes-shift between the excitation and the emission, which exceeds the LO phonon energy of about 300 cm−1 usually found for CdS nanocrystals cannot be assigned to dark exciton ground state. Therefore, we can conclude that the major contribution to the relatively large width of the emission spectra comes from defects with different natures inside the CdS nanocrystals or in their surfaces together with a large size distribution as evidenced by TEM measurement. Another reason for the significant broadening of this emission may come from the coupling of the electronic transitions with lattice phonons. Indeed, the strength of phonon coupling for DAP depends on both the distance between the donor and the acceptor centers and the localization of charges around them [69]. On the other hand, the appearance of a significant emission with an excitation wavelength of 532 nm, close to the bulk band gap (2.4 eV) even for low temperature and short annealing time (Fig. 7), is another proof of the great size of the nanocrystals.
Furthermore, the asymmetrical shape of the emission band can be convoluted at least with two Gaussian’s lines in accordance with the two bands, which appear in all the PL spectra near 730 and 790 nm respectively (Fig. 8). These bands may be assigned to two different populations of DAPs. Indeed, the energy of DAP depends on the distance separation between donor and acceptor according to the equation [70]:
where E g is the band gap, while E D and E A are the activation energies of donor and acceptor, respectively. The last term is the coulombic interaction energy between the donor and the acceptor, separated by a distance r and ε is the dielectric constant. Therefore, DAPs with small distance between donor and acceptor are responsible for the high-energy part of the emission band, whereas DAPs with larger separation emit at longer wavelengths. However, because of the great dispersion of donor–acceptor separation, the emission lines overlap forming a broad spectrum. Additionally, we have found that the ratio between the area of the high-energy band and that of the low-energy one decrease until it stabilizes at almost 4.6 indicating a dynamic evolution of the defects populations in the first few minutes of the growth. This result may be due to the excessive loss of sulfur, which induces an increase of sulfur vacancies and related defects as the time or the heat-treatment increases.

Photoluminescence spectrum convoluted by two Gaussian curves
We note however that the peak corresponding to the direct exciton recombination, usually seen in the high-energy region, was not observed in the emission spectra of our glass samples even for small particles and high excitation energy (355 nm). Indeed, many authors reported similar trends. According to Woogon [71], this is a common feature for materials prepared without special optimization, whereas other authors [48, 52] proposed different reasons for this behaviour. Santhi et al. [48] claimed that the absence of such emission might be due to a strong coupling between the nanoparticles and the glass matrix or to the nature and the quantity of traps. Weller [52] suggested that the absence of such peak is due to the formation of amorphous crystals. By capping CdS with sulfur, Gorer et al. [72], observed a prominent exciton emission peak and a weaker trap luminescence band. Indeed, band edge luminescence and deep-traps luminescence are interconnected since they involve the same defects. Therefore, great amount of deep traps may result in an extinction of the emission related to shallow ones. Moreover, the PL spectra, in our case, were recorded at room temperature. In these conditions, the luminescence arising from exciton recombination may be inefficient and too small for reasonable detection. In fact, at elevated temperatures, thermal occupation of phonon modes enhances the non-radiative decay rate as well as the emission stemming from trapping states.
Time-Resolved PL
To get further information about the states involved in fluorescence of this glass system, time resolved luminescence were carried out for different samples. As an example, the emission decay detected at various wavelengths near the maximum of the photoluminescence band is given in Fig. 9. According to this figure, the PL intensity revealed a non-exponential decay, which is in accordance with the heterogeneous character of the emitting centres. In fact, the occurrence of more than one decay time arises from the overlap between neighbour transitions with different relaxation mechanisms [52]. These transitions belong to the great variety of trapping centers in this kind of materials. However, we were able to fit the experimental PL decays with a bi-exponential function (Fig. 9). Table 4 shows the two decay times obtained from the fit of the experimental luminescence decay using the following equation:

Decay curves obtained at different emission wavelengths under excitation wavelength of 532 nm for the glass sample annealed at 365 °C for 10 min
In the above equation, I(t) is the time-dependent PL intensity, and τ1 and τ2 are the lifetimes of the two components with relative intensities A1 and A2. Since the obtained time constants far exceed the direct exciton recombination, we believe that the two components involved in the recombination process belong mainly to two sets of trapping states with different relaxation dynamics within the bandgap of the nanocrystal. According to Hache et al. [73], the slow component of the decay is due to surface states whereas the fast one is attributed to defects into the nanocrystal volume. Indeed, the transition probability as a function of donor–acceptor distance r is given by Thomas et al. [74]:
In this equation, ra is the Bohr radius of the donor, and W0, the limiting transition probability as r → 0, which is a constant for all pairs. Therefore, the wave function overlap between donor and acceptor dictate the time constants of particular DAP recombination; the more the donor and acceptor are closer, the more their lifetime is shorter. This statement may explain the decrease of the decay times with the particle size observed in this study (Table 4). Moreover, the recombination intensity of the DAP-radiation involves the pair distribution and the probability of pair occupation. Therefore, both size and composition may be responsible for the short-lived (τ1) and long-lived (τ2) components of fluorescence lifetime. Additionally, the two decay times vary with the emission wavelength (Table 5), which is also an indication of the contribution of different kinds of traps in the emission process. Consequently, not only the surface states, but also intrinsic defects inside the nanoparticles are equally important and can act as potential fluctuations to localize charge carriers during the recombination process. Besides, the nearly constant values of the parameters A1 and A2, determined by fitting Eq. 5 to the experimental emission decay, upon the average diameter of CdS crystallites (Table 4), indicate that the relative densities of traps are independent on the nanoparticle size and suggests that the emitting states have an intrinsic distribution [52].
In the light of the precedent arguments, we can draw the energy level diagram represented in Fig. 10, for CdS nanoparticles. In this model, the trapping states may arises from the defects, which were made during the sample preparation process or resulted from the laser irradiation. Accordingly, we may classify these defects into two different populations; those located within the inner shells and less influenced by the laser exposure and those situated at the nanocrystal surface and more sensible to the excitation power. Indeed, surface traps can also act as active sites for photochemical degradation of nanocrystals [75]. According to this scheme, excited carriers in the conduction band are rapidly trapped in surface or bulk defect states leading to trapping states. After that, recombination of photo generated-trapped electrons on Vs or ICd centers with holes trapped on VCd or IS sites, gives rise to the observed luminescence spectra. This suggestion is supported by the appearance of the red emission even for low delays, which indicates that the traps are very attractive to charge carriers [53].

Band diagram model illustrating the trap levels within the bandgap involved in the photoluminescence process
Conclusion
XRD and TEM investigation revealed the growth of CdS nanoparticles induced by the annealing of phosphate glass system. Optical absorption study revealed that particle growth is very fast in this kind of materials. From PL investigation, we concluded that the emission involves traps, which may be created during the growth process or under excitation light exposure. These traps were associated to sulfur or cadmium defects inside the nanocrystals and on their surfaces. We think that this CdS doped glass, which produces relatively intense emission extending from 600 to 850 nm, and a large Stokes shift may be used to produce LSCs with lower re-absorption and, hence, longer propagation lengths of luminesced photons to be guided to photovoltaic cell. Additionally, considering their suitable band gap for the visible light absorption, these CdS quantum dot doped glasses may be suitable for photocatalytic activity for H2 generation and dye degradation under visible light irradiation. Finally, these red and NIR emitting particles can provide a new class of luminophores with both long wavelength emission and long decay times. Therefore, this CdS doped glass may be of a considerable interest to achieve tunable broadband emission, which can be used as the gain medium of broadband fiber amplifiers and tunable fiber lasers emitting in the red region.
References
A. I. Ekimov and A. A. Onushchenko (1982). Sov. Phys. Semicond. USSR 16, 775–778.
A. Al Efros and A. Al Efros (1982). Sov. Phys. Semicond. 16, 772–775.
L. E. Brus (1984). J. Chem. Phys. 80, 4403–4409.
A. I. Ekimov, L. Al Efros, and A. A. Onushchenko (1985). Solid State Commun. 56, 921–992.
J. Warnock and D. D. Awschalom (1985). Phys. Rev. B 32, 5529–5531.
A. Henglein (1989). Chem. Rev. 89, 1861–1873.
G. T. Einevoll (1992). Phys. Rev. B 45, 3410–3417.
A. L. Efros, R. Rosen, M. Kuno, M. Nirmal, D. J. Norris, and M. Bawendi (1996). Phys. Rev. B 54, 4843–4856.
U. E. H. Laheld and G. T. Einevoll (1997). Phys. Rev. B 55, 5184–5204.
S. V. Nair and T. Takagahara (1997). Phys. Rev. B 55, 5153–5170.
R. Banerjee, R. Jayakrishnan, and P. Ayyub (2000). J. Phys.: Condens. Matter 12, 10647–10654.
H. Yu, J. Li, R. A. Loomis, L.-W. Wang, and W. E. Buhro (2003). Nat. Mater. 2, 517–520.
H. Hakan Gürel and H. Ünlü (2013). Mater. Sci. Semicond. Proc. 16, 1619–1628.
B. B. Kale, J.-O. Baeg, S. K. Apte, R. S. Sonawane, S. D. Naik, and K. R. Patil (2007). Mater. Chem. 17, 4297–4303.
Brenda C. Rowan, Lindsay R. Wilson, and Bryce S. Richards (2008). IEEE J. Sel. Top. Quantum Electron. 14, 1312–1322.
B. Girginer, G. Galli, E. Chiellini, and N. Bicak (2009). Int. J. Hydrog. Energy 34, 1176–1184.
S. K. Apte, S. N. Garaje, M. Valant, and B. B. Kale (2012). Green Chem. 14, 1455–1462.
N. Bel Haj Mohamed, M. Haouari, Z. Zaaboub, M. Nafoutti, F. Hassen, H. Maaref, and H. Ben Ouada (2014). J. Nanopart. Res. 16, 2242–2258.
F. Meinardi, A. Colombo, K. A. Velizhanin, R. Simonutti, M. Lorenzon, L. Beverina, R. Viswanatha, V. I. Klimov, and S. Brovelli (2014). Nat. Photon. 8, 392–399.
S. Halivni, S. Shemesh, N. Waiskopf, Y. Vinetsky, S. Magdassi, and U. Banin (2015). Nanoscale 7, 19193–19200.
N. Ben Brahim, N. Bel Haj Mohamed, M. Echabaane, M. Haouari, R. Ben Chaâbane, et al. (2015). Sens. Actuators B Chem. 220, 1346–135320.
N. Soltani, E. Saion, M. Z. Hussein, M. Erfani, A. Abedini, G. Bahmanrokh, M. Navasery, and P. Vaziri (2012). Int. J. Mol. Sci. 13, 12242–12258.
M. S. de la Fuente, R. S. Sánchez, V. González-Pedro, P. P. Boix, S. G. Mhaisalkar, M. E. Rincón, J. Bisquert, and I. Mora-Seró (2013). J. Phys. Chem. Lett. 4, 1519–1525.
E. V. Kolobkova (2006). Glass Phys. Chem 32, 404–411.
A. A. Lipovskii, E. V. Kolobkova, I. E. Yakovlev, N. V. Nikonorov, V. D. Petrikov, and A. A. Sitnikova (1997). J. Non Cryst. Solids 2, 2118–2645.
M. Elisa, C. Vasiliu, J. Striber, D. Radu, J. H. Trodahl, and M. Dalley (2006). J. Optoelectron. Adv. Mater. 8, 811–814.
Moonsub Shim, Congjun Wang, and Philippe Guyot-Sionnest (2001). J. Phys. Chem. B 105, 2369–2373.
V. L. Colvin, K. L. Cunningham, and A. P. Alivisatos (1994). J. Chem. Phys. 101, 7122–7138.
L. C. L. Amiot, S. Xu, S. Liang, L. Pan, and J. Xiaojun Zhao (2008). Sensors 8, 3082–3105.
F. Meinardi, A. Colombo, K. A. Velizhanin, R. Simonutti, et al. (2014). Nat. Photon. https://doi.org/10.1038/nphoton.2014.54.
B. Vaynberg, M. Matusovsky, M. Rosenbluh, et al. (1996). Opt. Commun. 132, 307–310.
E. V. Kolobkova, A. A. Lipovskii, and N. V. Nikonor (1995). Phys. Stat. Sol. (a) 147, K65.
A. M. Malyarevich, K. V. Yumashev, and A. A. Lipovskii (2008). J. Appl. Phys. https://doi.org/10.1063/1.2905320.
Yoshiyuki Asahara (1997). Ceram. Int. 23, 375–382.
A. Stanculescu, F. Stanculescu, and M. Elisa (2007). J. Optoelectron. Adv. Mater. 9, 828–835.
R. S. Sonawane, S. K. Apte, S. D. Naik, D. B. Raskar, and B. B. Kale (2008). Mater. Res. Bull. 43, 618–624.
D. Kim, C. Hwang, D. Gwoo, T. Kim, Y. Kim, and N. Kim (2011). Electron. Mater. Lett. 7, 309–312.
S. Wageh, A. M. El-Nahas, A. A. Higazy, and M. A. M. Mahmoud (2013). J. Alloy. Compd. 555, 161–168.
J. M. de Souza, D. N. Messias, V. Pilla, A. C. A. Silva, N. O. Dantas, and A. A. Andra (2017). Opt. Eng. 56, 121909.
D. A. Rayan, Y. H. Elbashar, M. M. Rashad, and A. El-Korashy (2013). J. Non Cryst. Solids 382, 52–56.
L. Al Efros and M. Rosen (2000). Annu. Rev. Mater. Sci. 30, 475–521.
B. Harieche, B. Boudine, O. Halimi, A. Fischer, A. Boudrioua, and M. Sebais (2011). J. Optoelectron. Adv. Mater. 13, 693–696.
A. Stanculescu, F. Stanculescu, and M. Elisa (2007). J. Optoelectron. Adv. Mater. 9, 828–835.
A. Lipovskii, E. Kolobkova, V. Petrikov, I. Kang, A. Olkhovets, Q. Shen, and S. Kycia (1997). Appl. Phys. Lett. 71, 3406–3408.
J. P. Poole (1949). J. Am. Ceram. Soc. 32, 230–233.
A. Chahboun, A. G. Rolo, S. A. Filonovich, and M. J. M. Gomes (2006). Sol. Energy Mater. Sol. Cells 90, 1413–1419.
Anke E. Abken, D. P. Halliday, and Ken Durose (2009). J. Appl. Phys. 105, 64515–64523.
S. Santhi, E. Bernstein, and F. Paille (2006). J. Lumin. 117, 101–112.
S. F. Wuister and A. Meijerink (2003). J. Lumin. 102, (103), 338–343.
A. E. Saunders, A. Ghezelbash, P. Sood, and B. A. Korgel (2008). Langmuir 24, 9043–9049.
S. F. Wuister and A. Meijerink (2003). J. Lumin. 105, 35–43.
H. Weller, U. Koch, M. Gutierrez, A. Henglein, and B. Bunsenges (1984). Phys. Chem. 88, 649–656.
N. Chestnoy, T. D. Harris, R. Hull, and L. E. Brus (1992). J. Chem. Phys. 90, 3393–3399.
B. Liu, G. Q. Xu, and L. M. G. Z. Shen (2001). J. Appl. Phys. 89, 1059–1063.
K. Misawa, H. Yao, and T. Kobayashi (1991). Chem. Phys. Lett. 183, 113–118.
N. R. Mou Pal, P. Mathews, and X. Ma Santiago (2012). J. Nanopart. Res. 14, 916.
A. Veamatahau, B. Jiang, T. Seifert, S. Makuta, K. Latham, M. Kanehara, and T. Teranishi (2014). Phys. Chem. Chem. Phys. 17, 2850–2858.
Pavan K. Narayanam, P. Purvesh Soni, R. S. Mohanta, S. S. Srinivasa, and S. S. Major Talwar (2013). Mater. Chem. Phys. 139, 196–209.
G. Q. Xu, B. Liu, S. J. Xu, C. H. Chewa, S. J. Chua, and L. M. Gana (2000). J. Phys. Chem. Solids 61, 829–836.
Katsumi Mochizuki, Masaaki Satoh, and Kenzo Igaki (1983). J. Appl. Phys. 22, 1414–1417.
V. Smyntyna, V. Skobeeva, and N. Malushin (2007). Radiat. Meas. 42, 693–696.
J. F. Suyver (2003). Synthesis, Spectroscopy and Simulation of Doped Nanocrystals. Thesis Debye Institute, Utrecht University, Nederland.
X. S. Zhao, J. Schrœder, P. D. Persans, and T. G. Bilodeau (1991). Phys. Rev. B 43, 12580–12589.
A. A. Lipovskii, E. V. Kolobkova, and V. D. Petrakos (1998). J. Cryst. Growth 184, (185), 365–369.
Nupur Saxena (2015). RSC Adv. 5, 73545–73551.
B. Capoen, A. Chahadih, H. El Hamzaoui, O. Cristini, and M. Bouazaoui (2013). Nanscale Res. Lett. 8, 266.
T. Miyoshi, K. Nitta, H. Ohkuni, F. Ikeda, and N. Matsuo (1997). Jpn. J. Appl. Phys. 36, 6726–6727.
J. Magyar, C. Aita, M. Gajdardziska-Josifovska, A. Sklyarov, K. Mikhaylichenko, and V. V. Yakovlev (2003). Appl. Phys. A 77, 285–291.
A. L. Gurskii and S. V. Viotikov (1999). Solid State Commun. 112, 339–343.
X. Xu, Y. Zhao, E. J. Sie, et al. (2011). ACS Nano 5, 3660–3669.
U. Woggon Optical Properties of Semiconductor Quantum Dots, Springer Tracts in Modern Physics, vol. 136 (Springer, Berlin, 1997).
S. Gorer, J. A. Ganske, J. C. Hemminger, and R. M. Penner (1998). J. Am. Chem. Soc. 120, 9584–9593.
F. Hache, M. C. Klein, D. Ricard, and C. Flytzanis (1991). J. Opt. Soc. Am. B 8, 1802–1806.
D. G. Thomas, J. J. Hopfield, and W. M. Augustiniak (1965). Phys. Rev. 140, A202–A220.
K. Sato, S. Kojima, S. Hattori, T. Chiba, K. Ueda-Sarson, T. Torimoto, Y. Tachibana, and S. Kuwabata (2007). Nanotechnology 18, 465702–465710.
Acknowledgements
The author would like to acknowledge Professors B. Champagnon and G. Panczer, Laboratoire de Physico-Chimie des Matériaux Luminescents, UMR 5620 CNRS, 12 Rue Ampère, Université Lyon 1, Villeurbanne, 69622 (France) for PL measurements facility.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest to declare.
Rights and permissions
About this article

Cite this article
Haouari, M., Saad, N. On the Origin of the Large Stokes-Shift of the Emission of CdS Nanoparticles Embedded in a Phosphate Glass Matrix. J Clust Sci 29, 391–402 (2018). https://doi.org/10.1007/s10876-017-1322-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10876-017-1322-x